Introduction
Global cancer statistics showed that there were approximately
2.2 million new cases of female Breast Cancer (BCa) and 684,996 associated deaths in 2020 [1]. BCa is the most common neoplasm among women and the leading cause of cancer in related morta- lities [1]. The hypernym BCa describes a diverse heterogeneous tumour group with a highly variable treatment response. Interes-tingly, TNBC, a subtype of BCa, accounts for approximately 1 in 5 cases of BCa but is disproportionately responsible for all BCa- associated deaths [2]. Clinically, TNBC is defined by its lack of ex- pression of the ER, PR, and HER2 receptors which are used as es- sential indicators to determine the optimal therapeutic protocols [3]. As reviewed by Yadav et al. patients with hormone dependent positive BCa benefit from chemo-targeted and hormone therapy, whereas treatment for TNBCs are currently limited to surgery and conventional cytotoxic agents [4].
In comparison to other BCa subtypes, TNBC tumours are fre- quently larger, less differentiated, and have a higher incidence of distant metastases, proliferation and recurrence [2,5]. TNBC is dif- ficult to manage, with the disease characterised by poor prognosis due to limited treatment options and subsequent drug resistance [6]. Notwithstanding the current advances in cancer chemothe- rapy, the ability of cancer cells to become chemo-resistant conti- nues to be a major obstacle in treating TNBC patients - especially in the metastatic setting [6]. Numerous mechanisms can lead to the development of chemoresistance, and the kisspeptin/kiss- peptin 1 receptor (KP/KISS1R) is a recognised system in the inves- tigation of TNBC drug-resistance (Figure 2) [7]. Thereby, there is an intense interest for novel agents which may synergise with cur- rent chemotherapeutic options and, consequently, attenuate the signalling potential of KISS1R. Currently, there are no small mole- cule KISS1R inhibitors reported against TNBC chemotherapeutic desensitisation, despite the evidence that the receptor may be a pharmaceutically attractive target for intervention against TNBC. In this paper, in addition to mapping the mechanistic pathway of KISS1/KISS1R expression, we explore the prospective implications of previously reported KISS1R antagonists and their effects in cur- tailing chemoresistance within TNBC.
KISS1R overview
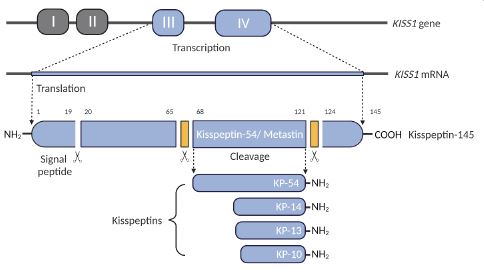
Kisspeptin receptor 1 (KISS1R; aka GPR54, OT7T175, AXOR12) is a Gαq/11-coupled G-Protein Coupled Receptor (GPCR) and a key regulator of the Hypothalamic-Pituitary-Gonadal (HPG) axis [8]. KISS1R is highly expressed in the brain, including the hypothala- mus and pituitary gland as well as peripheral regions [8]. Kisspep- tins (KPs), a product of the KISS1 gene, are a group of peptide frag- ments that bind to and activate the KISS1R (Figure 1). The KISS1 gene encodes a polypeptide consisting of 145 amino acids, known as the precursor peptide (KP-145) [9]. KP-145 gives rise to a se- cretory protein of 126 amino acids that is proteolytically cleaved into smaller fragments: KP-54 (aka Metastin), KP-14, KP-13 and KP-10 [8]. All peptide fragments bind to and activate the KISS1R with equal potency [10]. Each peptide fragment shares the same 10 terminal amino acid sequence, KP-10, the smallest fragment necessary for binding to and activating KISS1R [10]. The identi- fication of the KISS1 gene was initially championed for its anti- metastatic role, and KISS1activated KISS1R signalling has been shown to suppress cancer metastasis by inhibiting cancer cell migration and invasion [11]. Downregulation of KISS1 was clini- cally established with a worse prognosis among those diagnosed with melanoma [11], colorectal [12], prostate [13], ovarian [14], lung [15], and bladder cancers [16]. Paradoxically, elevated KISS1R signalling appears to play a pro-metastatic role in some cancers such as breast and liver cancer [17,18]. Successive findings have likened the metastases of TNBC basal-like malignancies, and sub- sequent drug resistance, with the overexpression of KISS1 [7]. As such, KISS1 expression might be a useful predictive biomarker in medical outcomes.
Role of KISS1R in TNBC development
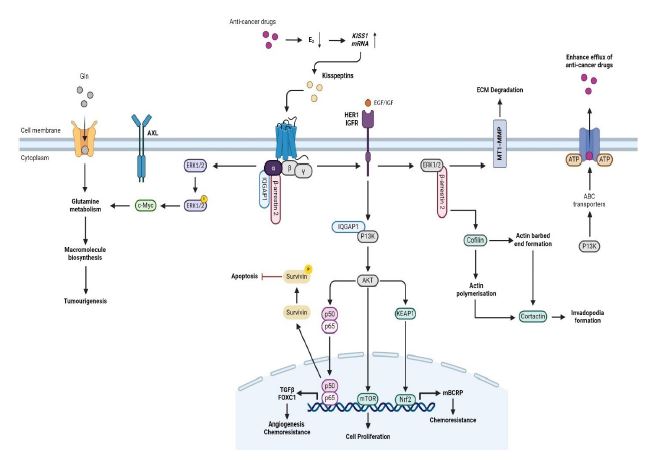
KISS1R signals through a plethora of diverse molecular mecha- nisms that have the potential to regulate the processes navigating clinical diagnoses of TNBCs. The underlying mechanisms by which the KP/KISS1R system regulates tumourigenesis in TNBC has been reviewed (Figures 2). Successive findings have likened the metas- tases of TNBC malignancies, and consequent drug resistance, with the overexpression of KISS1R.
To understand the mechanisms by which KISS1R stimulates BCa metastasis, Goertzen et al. examined the effects of KISS1R on invadopodia formation [19]. Goertzen et al. showed that G- protein dependent KISS1R promotes TNBC cell invasion by regu- lating the formation of invadopodia which are actin-rich protru- sions that facilitate extracellular matrix degradation and cancer cell invasion [19]. Goertzen also demonstrated that KP-10 induced KISS1R signalling which, in turn, induced two key regulators of actin dynamics during invadopodia maturation - the dephospho- rylation of cortactin and cofilin - through the adapter protein β-arrestin2 [19]. Treatment with the KISS1R ligand KP-10 in highly invasive TNBC cell lines (MDA-MB-231 and Hs578T) increased ge- latin degradation by TNBC cells, indicating enhanced invadopodia formation and activity. KP-10 stimulation led to colocalisation of the invadopodia markers F-actin and cortactin at sites of gelatin degradation [19]. Accordingly, Blake and co-workers, demons- trated that overexpression of KISS1R, in SKBR3 FLAKISS1R cells, also increased actin stress fibre formation [7]. KISS1R signalling also stimulated phosphorylation of MT1-MMP, a membrane-type matrix metalloprotease that plays a critical role in ECM degra- dation at invadopodia [19]. Goertzen et al. showed that KISS1R localised to invadopodia structures in TNBC cells upon KP-10 sti- mulation where it colocalised with cortactin, MT1-MMP, and the KISS1R interacting protein β-arrestin2 [19]. Notably, β-arrestin2 is a key mediator of KISS1R’s effects on invadopodia, for depletion of β-arrestin2 inhibited KP-10 induced invadopodia formation and the dephosphorylation of cortactin and cofilin. Moreover, it was demonstrated that KISS1R-stimlated invadopodia formation occurred through β-arrestin2 and ERK1/2 dependent pathways. Inhibition of the ERK1/2 pathway blocked KP-10 induced invado- podia formation and MT1-MMP phosphorylation [19].
Pampillo et al. demonstrated that β-arrestin2, in addition to GRK2, are critical regulators of early KISS1R signalling events, including desensitisation and coupling to the ERK1/2 MAPK pa- thway [20]. The study used HEK-293 cells to investigate the mo- lecular regulation of KISS1R as well as genetically modified MDA- MB-231 cells, expressing lower levels of β-arrestin2 compared to controls [20]. GRK2 was found to stimulate the desensitisation of KISS1R in HEK 293 cells, and this occurred through a phospho- rylation-independent mechanism [20]. In the MDA-MB-231 cells, β-arrestin2 was required for KISS1R activation of the ERK1/2 si- gnalling pathway, suggesting β-arrestin2 mediates this aspect of KISS1R signalling in this breast cancer cell line [20].
Cvetković et al. investigated the role of kisspeptins and their cognate receptor, KISS1R, in BCa metastasis [21]. The study de- monstrated that KP-10 stimulation induces invasion of ER-nega- tive MDA-MB-231 breast cancer cells via transactivation of the Epidermal Growth Factor Receptor (EGFR, aka HER1). The paper reports that exogenous expression of KISS1R in ER-negative SKBR3 breast cancer cells was sufficient to trigger invasion and induce extravasation in vivo. In contrast, KP-10 failed to transactivate the EGFR or stimulate invasiveness in ER-positive MCF7 and T47D breast cancer cells, thus, suggesting that ER negatively regulates KISS1R dependent breast cancer cell migration, invasion, and EGFR transactivation [21]. IQGAP1, an actin cytoskeletal binding partner of KISS1R, was shown to regulate EGFR transactivation in breast cancer. Cvetković and co-workers found that KP-10 induced EGFR phosphorylation was inhibited in IQGAP1 depleted cells (MDA-MB-231), suggesting that IQGAP1 is required for KISS1R induced EGFR activation [21]. It was demonstrated that KISS1R colocalises with IQGAP1 - in lamellipodia - at the leading edge of motile cells, indicating a potential role for IQGAP1 in transducing KISS1R signals to the cytoskeleton. The signalling of KISS1R can promote an invasive phenotype and was found to be regulated by the ER status of the breast epithelial cells. KISS1R expression was negatively regulated by E2 signalling through ER [21]. Overall, the findings strongly indicate that KISS1R mediated EGFR transactiva- tion in breast cancer cells.
Blake et al. assessed the effect of KISS1R overexpression in TNBC cell lines, MDA-MB-231 and Hs578T, as well as non-mali- gnant MCF10A and breast cancer SKBR3 cells [7]. The results demonstrated that KISS1R overexpression promoted Epithelial- Mesenchymal Transition (EMT)-like events, leading to increased tumour cell migration and invasion. Moreover, the overexpres- sion of KISS1R in SKBR3 cells stimulated cell motility and scratch closure, independent of increased cell proliferation. Notably it was observed that overexpression of KISS1R in SKBR3 cells led to significant increases in the expression of pro-survival molecules AXL, AKT, ERK and the anti-apoptotic protein survivin [7]. Stimu- lation of TNBC MDA-MB-231 and Hs578T, which endogenously express KISS1R, with KP-10 led to an upregulation in AXL protein and mRNA expression. Conversely, ER-positive breast cancer cell lines, T47D and MCF7, which have minimal KISS1R expression, do not express AXL. Blake et al. found a significant increase in the ex- pression of AXL in TNBC patients, which positively correlated with KISS1 expression [7]. Immunofluorescence analysis of patient’s tumours revealed punctate distribution of KISS1, robust localisa- tion of KISS1R and AXL in tumour cells, and partial co-localisation in tumours and stromal cells. Depletion of KISS1R signalling in- hibited metastatic TNBC cell migration, invasion, and malignant transformation. The findings suggest that KISS1, KISS1R and AXL are upregulated in invasive breast cancer cells, compared to non- invasive or weakly invasive breast cancer cells [7].
Dragan et al. identified the role of KISS1R as a novel regulator of TNBC metabolism and metastasis [22]. The study demonstra- ted that KISS1R overexpression changes the metabolic profile of TNBC cells, making them more dependent on glutamine for sur- vival [22]. Metabolic analysis of human primary tumour biopsies revealed elevated levels of glutamate in tumours compared to ER-positive tumours. Dragan et al. also highlighted the overex- pression of the MYC oncogene in TNBC, which promotes EMT and stimulates glutoaminolysis, leading to glutamine as a bioenergetic substate [22]. As such, KISS1R induces the expression of c-Myc to promote a glutamine-dependent phenotype and increase glutamine uptake in tumours. KISS1R induced c-Myc expression was dependent on ERK1/2 signalling. Inhibition of glutaminase or knockdown of c-Myc attenuated the growth and migration of KISS1R-overexpressing cells, demonstrating the importance of KISS1R-mediated glutamine metabolism for TNBC tumour pro- gress [22].
The findings have important implications for understanding the molecular mechanisms underlying KISS1R function and for the development of potential therapeutic strategies targeting this receptor. Targeting the KP/KISS1R signalling axis could have thera- peutic potential for TNBC.
Role of KISS1R in the acquisition of chemoresistance in TNBC
There is some evidence to suggest, as established by Huijbregts et al. and, that regulation of 17β-estradiol (E2) mediates the ex- pression of KISS1 in MDA-MB-231 cells [23]. Thus, the depletion of E2, brought upon by chemotherapy, supresses the downregu- lation of KISS1 expression [24]. Treatment with E2 was shown to dissuade KISS1 mRNA as well as KISS1R protein levels. It should be noted that higher levels of KP/KISS1R are expressed, clinically, upon exposure to chemotherapy. Levels of mRNA KISS1 are upre- gulated in TNBC cells which has been implicated in the increased protein/ KISS1R signalling as a factor that may induce breast can- cer progression and chemoresistance [24].
IQGAP1 is a multidomain protein that scaffolds multiple signal- ling pathways [21]. The scaffold role of IQGAP1 is emerging as key in the P13K-AKT pathway [6]. In agreement, Pan et al. showed that overexpression of IQGAP1 enhances AKT activation [25]. Stu- dies have demonstrated that AKT is the key signal transduction protein that phosphorylates a triad of downstream effectors, na- mely, Nrf2, NF-κB and mTOR [6]. The P13K-AKT-mTOR (PAM), NF-
κB and KEAP1-Nrf2 pathways enhanced chemotherapeutic drug resistance via BCRP transcription, thus, inducing the ATP-Binding Cassette (ABC) transporter family [6].
Blake et al. demonstrated that KISS1R signalling promotes the expression of the drug eflux transporter, ABC-G member 2 (ABCG2, BCRP), in metastatic TNBC cell lines [7]. KISS1R expres- sion led to a reduction in the cellular accumulation of doxorubicin, indicating a potential mechanism of drug resistance. Notably, the increase in expression of KISS1R was correlated with a heightened expression levels of BCRP mRNA and protein compared to control cells, while pharmacological inhibition of KISS1R decreased BCRP mRNA levels. Additionally, cells expressing KISS1R in TNBC tumour biopsies also expressed BCRP, reinforcing the association that KISS1R signalling promotes drug resistance by upregulating BCRP expression [7]. Blake et al. also identified AXL as a signalling partner in the KISS1R pathway, leading to increased signalling via AKT, and possibly regulating BCRP expression via an AXL/EMT-de- pendent mechanism [7]. The significant expression of AXL in TNBC patients, which positively correlated with KISS1 expression, may confer drug resistance in TNBC patients, as AXL overexpression was linked to poor TNBC patient prognosis, and has been shown to promote breast cancer drug resistance [7].
KISS1R expression plays a cardinal role in breast cancer resis- tance to chemotherapy. Drug resistance and tumourigenesis in TNBC, via KISS1R, can occur through various mechanisms (eflux drug transporter, BCRP, promotion of AXL expression and activity, PAM, NF-κB and KEAP1-Nrf2 pathways) including altered expres- sion and activation of KISS1R signalling, interaction with other signalling pathways, and genetic alteration affecting KISS1R func- tion.
Antagonistic agents: inhibition of KISS1R expression
Attenuation of KISS1R signalling via peptidic antagonists
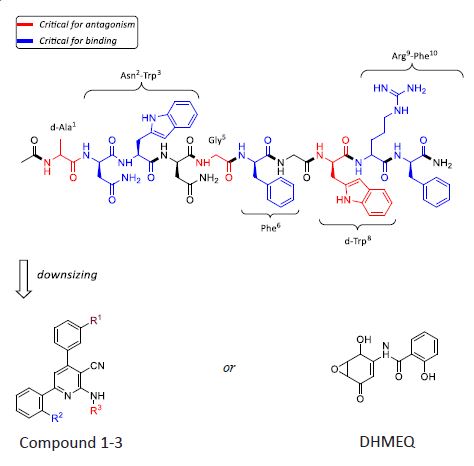
Several preclinical studies have indicated that KISS1R inhibition or KISS1R stimulation can be reliant on its cognate KPs. KP analo- gues have been synthesised based on the structure of human KP- 10, as this is the smallest fragment needed to bind to and activate KISS1R. In general, the antagonistic efficacy of KP-10 analogues have been assessed upon their ability to regulate KP-stimulated PLC-β mediated Inositol Phosphate (IP) and intracellular calcium release [26]. KISS1R’s capacity to signal via G-protein indepen- dent pathways attracts many secondary effectors (Figure 2). In consequence, recent reports assess an antagonist’s value upon its ability to inhibit downstream mediators [7,21,26]. Systematic substitution of amino-acids in KP-10, established by Roseweir et al., elucidated the functional potency of peptide-based KISS1R an- tagonists [26]. Four effective antagonists were created, whereby, valuable information regarding structural-activity relationship- derived pharmacophores and a consensus model for KP-10 antagonism were attained (Figure 3) [26]. Of those four antagonists, P-234 was the most efficacious, inhibiting KP-stimulated calcium release with an IC50 of 10 nM). In comparison, P-273 had a lower binding affinity than the decamer, KP-10, and P-230 displayed weak peptide-induced IP inhibition. Notwithstanding a complete reduction of KISS1R Ca2+ mobilisation, P-276 exhibited an idio- syncratic agonistic effect when administered at higher concen- trations. Thus, in vitro structure-activity data demonstrates that a KP amino-acid substitution from Tyr1, Ser5 and Leu8 to D-Ala1, Gly5 and D-Trp8 is critical for KISS1R antagonism. The data also purports that Asn2-Trp2, Phe6 and Arg9-Phe10 are the five essential amino acid residues involved in receptor binding [26].
Table 1: Reported KISS1R ligands: (i) amino-acid sequences of four key KISS1R peptidic antagonists; (ii) fluorescently labelled KP agonists.
| |
Name |
Sequence |
Reference |
| Agonist |
Kisspeptin-10 |
H-Tyr1-Asn2-Trp3-Asn4-Ser5-Phe6-Gly7-Leu8-Arg9-Phe10-NH2 |
[10] |
| TAK-448 |
Ac-d-Tyr1-Hyp2-Asn3-Thr4-Phe5-azaGly6-Leu7-Arg(Me)8-Trp9-NH2 |
[27,28] |
| Antagonist |
Peptide-230 |
Ac-d-Tyr1-Asn2-Trp3-Asn4-Gly5-Phe6-Gly7-Leu8-Arg9-Phe10-NH2 |
[26] |
| Peptide-234 |
Ac-d-Ala1-Asn2-Trp3-Asn4-Gly5-Phe6-Gly7-d-Trp8-Arg9-Phe10-NH2 |
| Peptide-273 |
Ac-d-Ala1-Asn2-Trp3-Asn4-d-Ser5-Phe6-Gly7-d-Trp8-Arg9-Phe10-NH2 |
| Peptide-276 |
Ac-d-Ala1-Asn2-Trp3-Asn4-Gly5-Phe6-Gly7 -Leu8-Arg9-Phe10-NH2 |
Based on its structure-activity profile and therapeutic poten- tial, P-234 has become the de-facto agent in mediating KP/KISS1R expression in various in vivo and in vitro systems [7,19,26]. In TNBC, the role of P-234/KISS1R signalling has not been fully elu- cidated, although studies have confirmed how inhibition of en- dogenously expressed KISS1R cells significantly attenuate TNBC tumourigenesis.
As demonstrated by Goertzen et al., KISS1R stimulates invado- podia formation via the co-localisation of actin and cortactin [19]. Moreover, they observed how down-stream mediators – namely, β-arrestin2 and ERK1/2 - are implicated in MT1-MMP phospho- rylation. Goertzen et al. showed how P-234 mediated KISS1R at- tenuation may impede tumour cell invasion, extra-cellular mem- brane degradation and metastasis. The KISS1R antagonist, P-234 inhibited KP-10 induced invadopodia formation demonstrating the specificity of KISS1R in regulating this process [19].
KISS1R’s capacity to promote drug resistance, as assessed in vitro by Blake et al., is mediated by numerous key effectors [7]. Nonetheless, Blake’s comprehensive insight in the downstream effects of KISS1R overexpression heighten the antagonistic effects of P-234. The researchers found that SKBR3FLAG-KISS1R cells dis- played increased cell survival in the presence of the chemothe- rapy drug doxorubicin, compared to control cells [7]. When the KISS1R antagonist, P-234 was used to attenuate KISS1R activity, the dose response curve shifted to the left, indicating increased cell death, similar to the response observed in control cells. This effect was also observed in other cell lines, including MCF10A breast cells expressing KISS1R and metastatic TNBC MDA-MB0231 cells. From the data provided by Blake et al. P-234 was shown to attenuate chemotherapeutic drug desensitisation and drug eflux (BCRP; Figure 2), up-regulation of pro-survival pathways (survivin, AXL, ERK, AKT; Figure 2), increase in HER1 transactivation, and an increase in TNBC cell motility [7].
Even so, therapeutic use of the known receptor ligands, such as P-234, would be complicated because they would need to be given parenterally, and stability of peptides are problematic be- cause KPs have been shown to degrade by the action of serum proteases [26]. As such, peptides may degrade over the course of the induction period such that the actual concentration of the an- tagonist is diminished across time. Hence, mimics of P-234 needs to avoid such shortcomings.
Following on from the pharmacophore model, developed by Roseweir et al. [26], we can begin to identify non-peptidic modu- lators of KISS1R, as potential drug candidates, as discussed below.
Attenuation of KISS1R signalling via small molecular antago- nists
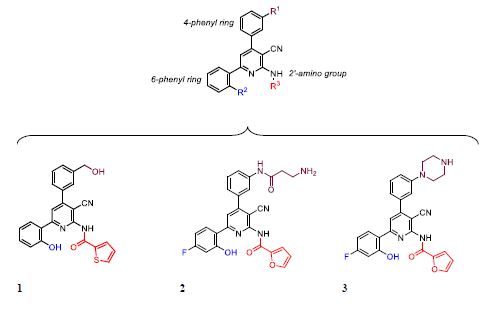
Kobayashi et al., identified compound 1 (Figure 4) as their most potent antagonist (58 % Ca2+ inhibition) of KISS1R-metastin signal- ling within the HPG axis via a high-throughput screen of their proprietary compounds [29]. They established a small molecular, non-peptide-based, antagonist based on a 2-acylamino-3-cya- no-4,6-diphenylpyridine scaffold, wherefrom a structure-activity relationship of the 2-amino group, 4-phenyl and 6 phenyl rings were founded. In their investigations of this pyridine scaffold, in- troduction of para-substituted or ortho-substituted hydroxy ace- tophenones, within a four component synthetic methodology, identified that a 2’-postion hydroxy group on the 6-phenyl ring was essential for the binding to KISS1R [29]. Moreover, introduc- tion of an electron-withdrawing group on the 4’-position of the 6-phenyl ring improved potency in Kobayashi’s series. Initially, they revealed how 2-acylamino-3-cyano-4,6-diphenylpyridine derivatives containing a primary amine on the 3’-subsituent on the 4-phenyl ring, and the presence of a 2-furoyl group, gave rise to the most active antagonist of all derivatives tested (compound 2) [29]. Thereafter, in a strategy to enhance Blood-Brain-Barrier (BBB) penetration, the primary amine on compound 2 was re- placed with a cyclic amine to afford antagonist, 3 (IC50 of 0.93 µM) [30]. Although compound 3 showed encouraging levels of KISS1R binding affinity in vitro, as well as some limited activity after do- sing in vivo, compound 3 did not antagonise calcium release to the same extent as P-234 [30].
Lin et al. examined the anti-cancer activity of the NF-κB in- hibitor Dehydroxymethylepoxyquinomicin (DHMEQ) on mouse plasmacytoma SP2/0 cells [31]. To investigate the role of KISS1R, Lin et al. performed knockdown experiments in SP2/0 cells using KISS1R targeted siRNA [31]. The KISS1R knockdown was found to suppress the cellular invasion of SP2/0 cells, suggesting that KISS1R plays an essential role in the invasion of these plasmacy- toma cells. Moreover, the inhibitory effect of DHMEQ on inva- sion was not further enhanced by KISS1R knockdown, indicating that the suppression of KISS1R expression is a key mechanism by which DHMEQ inhibits the invasive behaviour of SP2/0 cells. In addition to DHMEQ’s anti-invasive effects, the study also demons- trated that DHMEQ was able to enhance the cytotoxicity of the chemotherapeutic agent melphalan in SP2/0 cells. DHMEQ syner- gistically increased melphalan induced apoptosis, as evidenced by elevated levels of cleaved caspase-3. Further analysis showed that DHMEQ inhibited the expression of several NF-κB dependent anti- apoptotic proteins [31].
Conclusion and future outlook
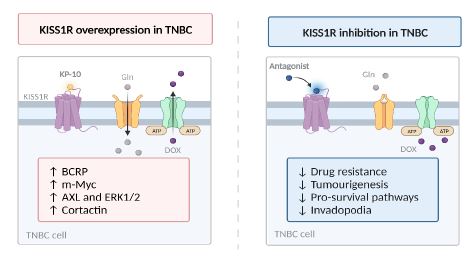
Characterised by poor survival rates, high instance of distance metastases, and limited therapeutic options, TNBC is the most aggressive form of BCa [1]. Thus, there is a particular medical in- terest to elucidate the molecular drivers behind TNBC chemore- sistance. KISS1R signals through a plethora of diverse molecular mechanisms that have the potential to regulate the processes na- vigating TNBC clinical outcomes [32]. There is growing evidence to support the notion that KISS1R plays a cardinal role in the late- stage progression of malignant, solid-tumour progression. A sum- mary of the significance of KISS1R overexpression and inhibition in TNBC is shown in Figure 5.
While further studies are required, to expand the structure- activity relationships around families of small, non-peptidic com- pounds, the data outlined in this review purports that substrates such as, Compounds 1-3 and (-) DHMEQ, could feasibly be de- veloped into alternatives to existing peptide-based KISS1R anta- gonists. Dissimilar to its peptide counterpart, which needs to be administered by injection, small molecule antagonists are more likely to be orally active and would therefore be more amendable to patient compliance in the clinic [29-31].
Going forward, the principal objective should be to evaluate the viability of KISS1R antagonism, as a therapeutic option in TNBC. We are highly engaged in this area and will report on our results with novel small molecule structure-activity relationships in due course [33]. Initially, more highly selective and potent KISS1R inhibitors should be developed to obtain better tolerability and anticancer efficacy. The question of whether KISS1R antago- nists can reduce angiogenesis and hence stop TNBC cells sprea- ding should also be addressed in the future [34,35]. In addition, a study around Kisspeptin regulating cell invasion and migration in endometrial cancer has been reported [36], which raises the need for an effect on cancer cell invasion and migration to be demons- trated with K1SS1R antagonists.
Secondly, optimised combination regimens, with existing che- motherapeutics (i.e., doxorubicin), need to be investigated for the best synergic effect. This will help to determine if KISS1R an- tagonists can effectively restore sensitivity to patients receiving established chemotherapies (TNBC often develops resistance to chemotherapy). Also, the prospect of utilising KISS1R antagonists in combination with standard chemotherapeutic agents, such as anthracyclines and taxanes, to potentially enhance the efficacy of these drugs is an enticing research area. Thirdly, TNBC is charac- terised by a subpopulation of cancer stem cells that contribute to recurrence and resistance to therapy. KISS1R antagonists may have the potential to inhibit the renewal and maintenance of can- cer stem cells, thereby reducing the likelihood of recurrence and improving long-term survival rates. It may also be worth investi- gating the use of KISS1R antagonists as an adjuvant therapy post- surgery, i.e. is it possible to eliminate residual cells and reduce chance of recurrence (something that is high in TNBC patients)? Moreover, KISS1R inhibitor resistance should also be investigated along with a greater understanding of the mechanism by which TNBC cells acquire drug resistance.
Additional studies should be conducted to identify reliable biomarkers, to predict antagonistic response and to minimise any adverse effects. Finally, can KISS1R expression levels serve as a biomarker to predict response to KISS1R antagonist therapy? Patients with higher KISS1R expression might benefit more from targeted therapy, i.e. more personalised clinical treatment. KiSS- 1 expression has been reported as a potentially useful prognostic marker in gastric cancer, and the hypothesis that this could be applied to breast cancer is worthy of future study [37].
Abbreviations: ABC: ATP Binding Cassette; ABCG2: ATP Binding Cassette G2; AKT: Protein Kinase B; ATP: Adenosine 5’ Triphos- phate; BBB: Blood-Brain Barrier; Bca: Breast Cancer; BCRP: Breast Cancer Resistant Protein; C-Myc: Cellular Myelocytomatosis; DAG: Diacylglycerol; E2: 17β-Estradiol; ECM: Extracellular Matrix; EFG: Epidermal Growth Factor; ER: Oestrogen Receptor; ERK1/2: Extracellular Signal-Regulated Kinases 1 and 2; GDP: Guanosine Diphosphate; Gln: Glutamine; GPCR: G-Protein Coupled Receptor; HER1: Human Epidermal Growth Factor Receptor 1; HER2: Human Epidermal Growth Factor Receptor 2; HPG: Hypothalamic-Pitui- tary-Gonadal; IC50: Inhibitory Concentration (Half-Maximal); IP: Inositol Phosphate; IP3: Inositol Trisphosphate; IQGAP1: IQ Motif Containing Gtpase Activating Protein 1; KEAP1: Kelch-Like ECH-As- sociated Protein 1; KISS1R: Kisspeptin 1 Receptor; KP: Kisspeptin; MT1-MMP: Membrane Type 1 Matrix Metalloproteinase; Mtor: Mechanistic Target Of Rapamycin; Nrf2: Nuclear Factor-Erythroid Factor 2-Related Factor 2; P13K: Phosphoinositide 3-Kinases; PIP2: Phosphatidylinositol 4,5-Bisphosphate; PKC: Protein Kinase C; PLC-Β: Phospholipase C-Β; PR: Progesterone Receptor; TNBC: Triple-Negative Breast Cancer.
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Can- cer J Clin. 2021; 71(3): 209-49.
- Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, et al. Differences in breast carcinoma characteristics in newly diagnosed African- American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epide- miology, and End Results database. Cancer. 2007; 110(4): 876-84.
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, et al. Triple- negative breast cancer: clinical features and patterns of recur- rence. Clin Cancer Res. 2007; 13(15 Pt 1): 4429-34.
- Yadav BS, Sharma SC, Chanana P, Jhamb S. Systemic treatment strategies for triple-negative breast cancer. World J Clin Oncol. 2014; 5(2): 125-33.
- Freedman GM, Anderson PR, Li T, Nicolaou N. Locoregional recur- rence of triple-negative breast cancer after breast-conserving sur- gery and radiation. Cancer. 2009; 115(5): 946-51.
- Dong C, Wu J, Chen Y, Nie J, Chen C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front Pharma- col. 2021; 12: 628690.
- Blake A, Dragan M, Tirona RG, Hardy DB, Brackstone M, et al. G protein-coupled KISS1 receptor is overexpressed in triple nega- tive breast cancer and promotes drug resistance. Sci Rep. 2017; 7: 46525.
- Gottsch ML, Cunningham MJ, Smith JT, Popa SM, Acohido BV, et al. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004; 145(9): 4073-7.
- Castano JP, Martinez-Fuentes AJ, Gutierrez-Pascual E, Vaudry H, Tena-Sempere M, et al. Intracellular signaling pathways activated by kisspeptins through GPR54: do multiple signals underlie func- tion diversity? Peptides. 2009; 30(1): 10-5.
- Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vande- rwinden JM, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001; 276(37): 34631-6.
- Lee JH, Miele ME, Hicks DJ, Phillips KK, Trent JM, et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. 1996; 88(23): 1731-7.
- Okugawa Y, Inoue Y, Tanaka K, Toiyama Y, Shimura T, et al. Loss of the metastasis suppressor gene KiSS1 is associated with lymph node metastasis and poor prognosis in human colorectal cancer. Oncol Rep. 2013; 30(3): 1449-54.
- Wang H, Jones J, Turner T, He QP, Hardy S, et al. Clinical and biologi- cal significance of KISS1 expression in prostate cancer. Am J Pathol. 2012; 180(3): 1170-8.
- Cao F, Chen L, Liu M, Lin W, Ji J, et al. Expression of preoperative KISS1 gene in tumor tissue with epithelial ovarian cancer and its prognostic value. Medicine (Baltimore). 2016; 95(46): e5296.
- Zheng S, Chang Y, Hodges KB, Sun Y, Ma X, et al. Expression of KISS1 and MMP-9 in non-small cell lung cancer and their relations to me- tastasis and survival. Anticancer Res. 2010; 30(3): 713-8.
- Takeda T, Kikuchi E, Mikami S, Suzuki E, Matsumoto K, et al. Pro- gnostic role of KiSS-1 and possibility of therapeutic modality of metastin, the final peptide of the KiSS-1 gene, in urothelial carci- noma. Mol Cancer Ther. 2012; 11(4): 853-63.
- Ikeguchi M, Hirooka Y, Kaibara N. Quantitative reverse transcrip- tase polymerase chain reaction analysis for KiSS-1 and orphan G- protein-coupled receptor (hOT7T175) gene expression in hepato- cellular carcinoma. J Cancer Res Clin Oncol. 2003; 129(9): 531-5.
- Martin TA, Watkins G, Jiang WG. KiSS-1 expression in human breast cancer. Clin Exp Metastasis. 2005; 22(6): 503-11.
- Goertzen CG, Dragan M, Turley E, Babwah AV, Bhattacharya M. KISS1R signaling promotes invadopodia formation in human breast cancer cell via beta-arrestin2/ERK. Cell Signal. 2016; 28(3): 165-76.
- Pampillo M, Camuso N, Taylor JE, Szereszewski JM, Ahow MR, et al. Regulation of GPR54 signaling by GRK2 and beta-arrestin. Mol Endocrinol. 2009; 23(12): 2060-74.
- Cvetkovic D, Dragan M, Leith SJ, Mir ZM, Leong HS, et al. KISS1R in- duces invasiveness of estrogen receptor-negative human mamma- ry epithelial and breast cancer cells. Endocrinology. 2013; 154(6): 1999-2014.
- Dragan M, Nguyen MU, Guzman S, Goertzen C, Brackstone M, et al. G protein-coupled kisspeptin receptor induces metabolic repro- graming and tumorigenesis in estrogen receptor-negative breast cancer. Cell Death Dis. 2020; 11(2): 106.
- Huijbregts L, de Roux N. KISS1 is down-regulated by 17beta-estra- diol in MDA-MB-231 cells through a nonclassical mechanism and loss of ribonucleic acid polymerase II binding at the proximal pro- moter. Endocrinology. 2010; 151(8): 3764-72.
- Septiani RV, Soewoto W, Budhi IB. Chemotherapy Effect on Estra- diol Levels in Patients with Triple-negative Breast Cancer: A Clinical Prospective Study from Indonesia. Open Access Macedonian Jour- nal of Medical Sciences. 2022; 10(B): 477-81.
- Pan CW, Jin X, Zhao Y, Pan Y, Yang J, et al. AKT‐ phospho- rylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1‐ MAPK interaction. The EMBO Journal. 2017; 36(8): 995-1010-.
- Roseweir AK, Kauffman AS, Smith JT, Guerriero KA, Morgan K, et al. Discovery of potent kisspeptin antagonists delineate physiological mechanisms of gonadotropin regulation. J Neurosci. 2009; 29(12): 3920-9.
- Matsui H, Tanaka A, Yokoyama K, Takatsu Y, Ishikawa K, et al. Chro- nic administration of the metastin/kisspeptin analog KISS1-305 or the investigational agent TAK-448 suppresses hypothalamic pitui- tary gonadal function and depletes plasma testosterone in adult male rats. Endocrinology. 2012; 153(11): 5297-308.
- Matsui H, Asami T. Effects and therapeutic potentials of kisspep- tin analogs: regulation of the hypothalamic-pituitary-gonadal axis. Neuroendocrinology. 2014; 99(1): 49-60.
- Kobayashi T, Sasaki S, Tomita N, Fukui S, Kuroda N, et al. Synthe- sis and structure-activity relationships of 2-acylamino-4,6-diphe- nylpyridine derivatives as novel antagonists of GPR54. Bioorg Med Chem. 2010; 18(11): 3841-59.
- Kobayashi T, Sasaki S, Tomita N, Fukui S, Nakayama M, et al. 2-acy- lamino-4,6-diphenylpyridine derivatives as novel GPR54 antago- nists with good brain exposure and in vivo efficacy for plasma LH level in male rats. Bioorg Med Chem. 2010; 18(14): 5157-71.
- Lin Y, Sidthipong K, Ma J, Koide N, Umezawa K, et al. The designed NF-kappaB inhibitor, DHMEQ, inhibits KISS1R-mediated invasion and increases drug-sensitivity in mouse plasmacytoma SP2/0 cells. Exp Ther Med. 2021; 22(4): 1092.
- Cvetkovic D, Babwah AV, Bhattacharya M. Kisspeptin/KISS1R Sys- tem in Breast Cancer. J Cancer. 2013; 4(8): 653-61.
- Wren SP, et al. Unpublished results.
- Matjila M, Millar R, van der Spuy Z, Katz A. The differential expres- sion of Kiss1, MMP9 and angiogenic regulators across the feto- maternal interface of healthy human pregnancies: Implications for trophoblast invasion and vessel development. PLoS One. 2013; 8(5): e63574.
- Ly T, Harihar S, Welch DR. KISS1 in metastatic cancer research and treatment: potential and paradoxes. Cancer Metastasis Rev. 2020; 39(3): 739-754.
- Wu HM, Chen LH, Chiu WJ, Tsai CL. Kisspeptin Regulates Cell In- vasion and Migration in Endometrial Cancer. J Endocr Soc. 2024; 8(3): bvae001.
- Dhar DK, Naora H, Kubota H, et al. Downregulation of KiSS-1 ex- pression is responsible for tumor invasion and worse prognosis in gastric carcinoma. International Journal of Cancer. 2004; 111(6): 868-872.