Introduction
Gorlin Syndrome (GS), is an autosomal dominant familial cancer predisposition disorder. It is also known as Gorlin-Goltz syndrome, Basal Cell Nevus Syndrome (BCNS), or nevoid basal cell carcinoma syndrome [1]. It is a disorder characterized by multiple developmental abnormalities and an increased predisposition to various cancers. The first description of the distinct syndrome consisting of multiple nevoid basal cell epitheliomas, jaw cysts and bifid ribs was published in 1960 [2]. This multisystemic disease has a prevalence of 1 in 40,000-60,000 and affects men and women equally [3]. It has a high penetrance and variable expression. Although detected in young children, GS is most commonly expressed between the ages of 17 and 35 [4]. Several phenotypic characteristics may occur, some since childhood, such as multiple Basal Cell Carcinomas (BCCs); skeletal, ophthalmological, and neurologic alterations [5].
For the diagnosis of GS, 2 major criteria, 1 major criterion, and 2 minor criteria, or 1 major criterion plus molecular confirmation are required [6,7,8]. Major criteria are: multiple (>2) BCCs or 1 BCC under 20 years of age; histologically proven Odontogenic Keratocysts (OKCs) of the jaws; palmar or plantar pits (three or more); bilamellar calcification of the falx cerebri; bifid, fused or markedly splayed ribs; and first-degree relative with BCNS. More than 100 minor criteria have been also described [7].
Mutations in the Sonic Hedgehog (SHH) pathway genes, most notably PTCH1, cause this rare disease and are responsible for 50 to 85% of cases of GS. This tumor suppressor gene, located on the long arm of chromosome 9, encodes a transmembrane receptor capable of recognizing SHH signaling proteins. In particular, PTCH1 de novo mutations are responsible for 20 to 30 % of new cases of GS [8]. Mutations in the fused gene (SUFU) and, less commonly, in PTCH2 have also been reported [9].
A multidisciplinary approach is required to manage patients with GS. Adult patients should have a baseline MRI scan of the brain, genetic counseling, and preferably psychological aid. Patients with a history of medulloblastoma should have repeated X-rays of the jaw as needed and should have an annual neurological examination [10]. Avoidance of radiotherapy is strongly recommended for all patients with GS due to radiation hypersensitivity [11].
Treatment methods can be grouped as follows: surgical treatment, non-surgical treatment, and treatment based on molecular pathogenesis. SHH pathway aberrations are found in sporadic BCCs, which favors therapeutic agents targeting known key components of the signaling pathway. Vismodegib (Erivedge), also known as GDC 0449, a first-in-class SHH pathway inhibitor, was approved by the US Food and Drug Administration for BCC in 2013 [12]. It has been developed and tested in somatic or germline mutation disease carriers with advanced disease and can help in those cases in which radiation is contraindicated, or the lesions are inoperable. The most common adverse reactions are muscle spasms, alopecia, dysgeusia, weight loss, fatigue, nausea, diarrhea, decreased appetite, constipation, cough, arthralgias, vomiting, headache, ageusia, insomnia, and upper respiratory tract infection [13]. Primary or secondary resistance is seen in 20% of patients [14].
We reported the case of a GS patient with a VUS (Variant of Uncertain Significance) in the PTCH1 gene who fulfilled limited major and minor diagnostic criteria but ultimately 15 achieved a positive outcome after a targeted therapy.
Patient and methods
A 44-year-old male patient presented to the Medical-Oncology Department of the Hospital de Clínicas Academic Hospital in Montevideo, Uruguay, in January 2023, with a personal history of cleft lip and multiple BCCs.
Genetic testing for inherited germline mutations and deletion/duplication detection in the PTCH1 and SUFU genes has been carried out.
The researchers obtained informed consent from the participant to publish information and images.
The study was approved by the institutional review boards of the Military Hospital, Montevideo, Uruguay, and complied with the current version of the Declaration of Helsinki. Written informed consent was obtained during genetic counseling sessions.
Results
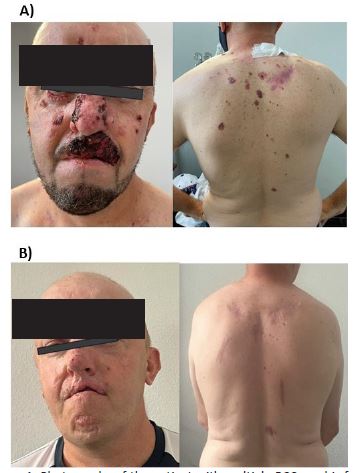
The 44-year-old male patient had large lesions that disfigured functionally sensitive areas such as the nose, the eyelid, and the lower and upper lip. Although he only fulfilled one major and one minor clinical criterion, it was considered suspicious for GS and offered genetic counseling at the Uruguayan Oncogenetic Centre. No relevant familial history was obtained.
Genetic testing revealed a VUS in the PTCH1 gene (17c.1846A>G p.Ser616Gly). It is not present in population databases and has not been previously reported in the literature in individuals affected with PTCH1-related conditions. Using insilico algorithms to predict sequence changes in RNA splicing, this variant could disrupt the consensus splice site.
According to consensus guidelines for locally advanced BCC [15], and based on high clinical suspicion and insilico evidence of PTCH1 protein dysfunction, the patient was ultimately offered targeted therapy. Then, oral treatment with vismodegib 150 mg/day was administered. In addition, some of the dorsal lesions were also surgically removed. After 12 months of follow-up, alopecia was the only adverse effect reported. Complete clinical remission was achieved (Figure 1).
Discussion
Gorlin-Goltz syndrome is an uncommon autosomal dominant inherited disorder mainly characterized by numerous BCCs, OKCs, and musculoskeletal malformations. Clinically manifesting with almost complete penetrance and variable phenotypic findings.
Our patient fulfilled only 1 major and 1 minor clinical criterion and had no family history of GS manifestations. He was an only child with no offspring, and both parents were deceased.
Genetic testing revealed a VUS in the PTCH1 gene. This gene was first isolated in 1996 as the human homolog of the Drosophila segment polarity PTCH1 gene, mapped in the long arm of chromosome 9q22.3-q31 [16]. It encodes a transmembrane protein that functions as a receptor for the SHH molecules. Mutations in the PTCH1 gene result in loss of control of several genes known to play a role in organogenesis, carcinogenesis, and odontogenesis.
Inhibitors of the SHH pathway have determined a shift for locally advanced and metastatic BCCs, especially for recurred or not eligible patients for surgery and/or radiation therapy [17].
Based on our results, all the data on the variant, the inheritance pattern, and the improvement in symptoms following targeted therapy, a reclassification of this variant may be considered.
Some limitations of this study included the absence of somatic testing. Regarding the patient family, due to the family composition and the deceased relatives, we were not able to evaluate the variant co-segregation and confirm or discard the assumption of a de novo variant.
Conclusion
In this paper we present a Uruguayan patient with GS. He has a probably de novo pathogenic variant with an incomplete phenotype. Molecular diagnosis was critical to the implementation of targeted therapy, resulting in a complete response with minimal adverse events. High clinical suspicion, early genetic diagnosis, and timely tailored treatment are essential to reduce the severity of complications, deformity, and malignant predisposition, favoring appropriate management based on targeted therapy.
References
- Hazemann G, Michel C, Mahé A, et al. Histopathological study of basaloid follicular hamartoma. Ann Dermatol Venereol. 2019; 146(3): 181-191.
- Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960; M262: 908-12.
- Spiker AM, Troxell T, Ramsey ML. Gorlin Syndrome. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Available from: https://www.ncbi.nlm.nih.gov/books/NBK430921/
- Ramaglia L, Morgese F, Pighetti M, et al. Odontogenic keratocyst and uterus bicornis in nevoid basal cell carcinoma syndrome: Case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006; 102: 217-9.
- Hasan A, Akintola D. An Update of Gorlin-Goltz Syndrome. Prim Dent J. 2018; 7(3): 38-41.
- Bree AF. Shah MR for the BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet Part A. 2011; 155: 2091-2097.
- Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993; (6): 460-4.
- Witmanowski H, Szychta P, Błochowiak K, Jundziłł A, Czajkowski R. Basal cell nevus syndrome (Gorlin-Goltz syndrome): Genetic predisposition, clinical picture and treatment. Postepy Dermatologiii Alergologii. 2017; 34(4): 381-387.
- Jawa DS, Sircar K, Somani R, Grover N, Jaidka S, et al. Gorlin Goltz syndrome. A case report. J Oral Maxillofac Pathol. 2009; 13: 89-92.
- Soufir N, Gerard B, Portela M, Brice A, Liboutet M, et al. PTCH mutations and deletions in patients with typical nevoid basal cell carcinoma syndrome and in patients with a suspected genetic predisposition to basal cell carcinoma: a French study. Br J Cancer. 2006; 95(4): 548-53.
- Onodera S, Nakamura Y, Azuma T. Gorlin Syndrome: Recent Advances in Genetic Testing and Molecular and Cellular Biological Research. Int J Mol Sci. 2020; 21(20): 7559.
- Bay C, Ousager LB, Jelsig AM. Patients with basal cell naevus syndrome should be offered an early multidisciplinary follow-up and treatment. Ugeskr Laeger. 2015; 177(29).
- Casey D, Demko S, Shord S, Zhao H, Chen H, et al. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017; 23(10): 2377-2381.
- Axelson M, Liu K, Jiang X, He K, Wang J, et al. U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin Cancer Res. 2013; 19: 2289-2293
- Jacobsen AA, Aldahan AS, Hughes OB, et al. Hedgehog Pathway Inhibitor Therapy for Locally Advanced and Metastatic Basal Cell Carcinoma: A Systematic Review and Pooled Analysis of Interventional Studies. JAMA Dermatol. 2016; 152(7): 816-24.
- Atwood SX, Chang ALS, Oro AE. Hedgehog pathway inhibition and the race against tumor evolution. J. Cell Biol. 2012; 199: 193-197.
- Peris K, Fargnoli MC, Garbe C, et al. Diagnosis and treatment of basal cell carcinoma: European consensus–based interdisciplinary guidelines. Eur. J. Cancer. 2019; 118: 10-34.
- Cohen MM. Jr Nevoid basal cell carcinoma syndrome: Molecular biology and new hypotheses. Int J Oral Maxillofac Surg. 1999; 28: 216-23.
- Peris K., Licitra L., Ascierto P.A., et al. Identifying locally advanced basal cell carcinoma eligible for treatment with vismodegib: An expert panel consensus. Future Oncol. 2015; 11: 703-712.