Introduction
Currently, cancer is among top 10 leading causes of death in the world. According to GLOBOCAN, 9.7 million people died from cancer worldwide in 2022, corresponding to 16,8% of deaths [1]. The disease was responsible for 30.3% of premature deaths in those aged 30-69 years in 2022 and is also closely related to a decrease in life expectancy, since 39.61% of cancer casualties occurred in the age range of 50-69 and 46.93% over 70 years old in 2019 [2].
Cancer can be defined as a genetic disease of multifactorial origin, in which cells acquire functional capacities that allow their abnormal proliferation, resistance to apoptosis and invasion of other tissues, characterizing a malignant neoplastic state with the formation of a mass tumor (solid malignant tumor), except in blood cancer [3].
There are more than 200 types of cancer, usually named by the site of origin and the cell type that formed it, with carcinomas being the most common [4]. In 2000, Hanahan and Weinberg summarized six functional capacities acquired by cells during carcinogenesis [5]. With the advancement of research in Oncology, the authors added eight more capabilities that can be acquired by cancer cells, demonstrating the high complexity of this disease [6,7]. Amongst them, tissue invasion and metastasis may occur in some types of malignant tumors during their tumor progression. Metastasis is recognized as the development of a secondary tumor distant from the site where the original primary tumor is located [8], requiring a series of dynamic processes for a competent heterogeneous population to leave the primary site, circulate and resist bloodstream pressure, adapt to a new environment, resist immune system attack, and colonize to form a secondary tumor [8,9]. Initially, cells undergo a process of transdifferentiation, in which they gain migratory and invasive properties by executing a multistep program known as Epithelial-Mesenchymal Transition (EMT) [10]. After EMT, cells need to survive in the peripheral or lymphatic circulation, showing resistance to apoptosis and chemotherapy [7]. With the survival of a competent population, these cells must follow the reverse path, in which they lose their migratory capabilities and settle and colonize the new site [11].
Many studies have reported a correlation between invasiveness and chemotherapy resistance, being metastatic disease the primary cause of cancer-related deaths [12-14]. In that context, ABC transporters are known to play a key role in cellular detoxification and excretion of noxious molecules and the simply overexpression of ABC transporters is associated with the development of the Multidrug Resistance (MDR) phenotype in cancer cells after prolonged exposure to chemotherapy drugs, consisting in a poor prognosis for disease treatment [14,15]. Considering the relationship between resistance and metastasis is not yet well clarified in the scientific literature, this review intends to bring to light the crucial findings in this field of investigation, contributing to the search for therapeutic targets that allow a more efficient chemotherapy in cases of resistant and metastatic cancers.
Drug resistance and ABC transporters
Drug resistance is the main obstacle to chemotherapy and is a phenomenon as old as the field itself. Cancer chemotherapy was boosted by the success of chemotherapy for infectious diseases; however, with the description of the toxic effect of a drug on microorganisms, resistant cells emerged from these cultures at the same time [16]. Since then, resistance has followed chemotherapy like a faithful shadow and the history of both are interconnected issues.
Articles on cancer chemotherapy and resistance began in 1944 by Heilman and Kendall, where they demonstrated that mice with lymphosarcoma became refractory to chemotherapy, showing cross-resistance to treatment with cortisone plus polyphenol phlorhizin, and cortisone plus another steroid estradiol propionate [17]. In 1950, Burchenal and colleagues selected a folic acid-resistant AK4 leukemia cell line after three to four passages in mice treated with amethopterin, also known as methotrexate [18]. This resistant strain called AK4R showed cross-resistance to folic acid analogues [19], but preserved cortisone sensitivity as the parental strain, though it showed collateral sensitivity to 2,6-diaminopurine [20]. In 1958, Fisher describes the first cultures of cells derived from the murine leukemias L5178 and P815 with continuous reproduction in vitro and with acquired resistance to methotrexate [21].
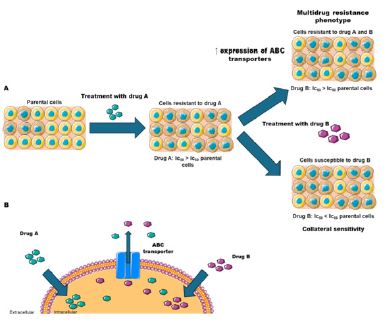
It is interesting to point out that resistance to chemotherapy may be of intrinsic/natural origin and/or acquired by prolonged exposure to chemotherapy. According to Hutchison (1963), cross-resistance means that a population resistant to a certain compound (for example, drug 1) is not susceptible to another compound (drug 2) while the parental population remains susceptible to both. Collateral susceptibility is defined as an increased susceptibility of a resistant population relative to the parent population to another compound (drug 3), see figure 1 for details [16].
Most metastatic tumors are resistant to chemotherapy and apoptosis (intrinsic resistance) or respond to chemotherapy initially, but soon a minute amount of resistant cells are selected by the treatment. This population is associated with the broad-spectrum resistance or MDR phenotype, characterized by cross-resistance to different functionally and structurally unrelated chemotherapeutics. Many mechanisms have been proposed to explain the MDR phenotype that may be associated with drug entry, metabolism and/or efflux [16]. In 1963, Adamson and co-workers demonstrated that resistant tumors of L1210 leukemia had one third of the radioactively labeled anti-leukemic agent NSC 32946 than sensitive tumors after 24 h of administration in mice [22]. Sivak et al. (1964) showed increased accumulation of the anti-leukemic drug NSC 38280 in vivo in strains derived from solid tumors and ascites of leukemia murine P388 and L1210 sensitive versus resistant, suggesting the existence of distinct mechanisms of transport, binding or metabolism of this phthalanilide derivative [23]. Furthermore, disposition studies suggested that NSC-38280 maintained its molecular integrity in vivo, ruling out the presence of a derived metabolite [23,24], raising the hypothesis of a drug efflux mechanism in these cells. Yesair and co-workers in 1966 found a substantial loss of the cancer chemotherapeutic agent NSC 60339 in P388 cells when washed with sucrose relative to saline [25]. Kessel et al (1968) presented the same suspicion regarding Daunorubicin (DNR) efflux in several leukemic cell lines cross-resistant to DNR, vinblastine and NSC 38280 and described a rapid loss of drugs in resistant cells previously loaded, which could be related to drug response [26]. A few years later, Dano(1973) demonstrated in his remarkable work that the efflux of DNR occurred against its concentration gradient and was energy-dependent and temperature-sensitive [27].
Analyzes carried out on a Chinese hamster ovary MDR cell line in 1976, performed by Juliano and Ling, identified a membrane protein of 170 KDa that conferred resistance against high concentrations of colchicine [28]. Due to thought related to alteration in cell permeability, it was firstly named P-glycoprotein (P for permeability), being later also termed MDR1 (multidrug resistance protein-1) and more recently denominated ABCB1 (ABC transporter, subfamily B, member 1) [29]. In 1985, Bell et al. detected overexpression of ABCB1 in patients with ovarian cancer and correlated this result with the development of the MDR phenotype [30].
Although ABCB1 was first described in cancer cells, it is also expressed in normal tissues, typically located on the apical surface of cells. The expression was found in many organs such as the brain, lung, gastrointestinal tract, testis, placenta, adrenal and kidneys, where it participates in the bioavailability of molecules and cell detoxification [31,32]. In humans, two genes belong to the MDR family: mdr1/abcb1 and mdr2/abcb4, which encode two homologous proteins, though only the first is related to the MDR phenotype. The two isoforms share 78% of the sequence identity and it is suggested that they present similar structures and mechanisms of action [33]. The protein has a structure typical of ABC proteins, expressed as a single polypeptide chain of 1280 amino acids, containing two homologous regions, each with a transmembrane domain and a nucleotide-binding domain, separated by a flexible linker region. ABCB1 can transport a wide variety of substrates, which vary in size, structure and function. Most are weakly amphipathic and relatively hydrophobic, usually containing aromatic rings and a positively charged nitrogen atom [34].
In 1992, the Multidrug Resistance Protein-1 (MRP-1) or ABCC1 (ABC transporter, subfamily C, member 1), was identified in lung tumor cell lines H69R resistant to anthracyclines, a class of key molecules in neoplastic chemotherapy [35]. The ABCC1 protein was the first of the ABCC subfamily to be identified out of a total of 13 members, of which nine are involved in the transport of drugs, called MRP [36]. The ABCC1 have a larger structure than the others ABCs, due to the presence of an extra domain with five transmembrane helices (MSD0), located in the N-terminal portion of the polypeptide chain of 1531 amino acids and 190 KDa [37]. The ABCC1 protein can mediate cellular efflux of a variety of physiological organic anions, xenobiotics and their metabolites. Many of these organic anions are conjugated to glutathione (GSH), glucuronide or sulfate; however, the protein can carry out transport without conjugation, or in cotransport with GSH [38]. ABCC1 is found in all organs and tissues, though its expression is relatively higher in certain defense tissues such as blood-brain, blood-testis, lungs, skin, intestine, kidney and placenta, consistent with a role in cellular detoxification [32,39].
In 1998, the ABCG2 transporter (ABC transporter, subfamily G, member 2) was identified by three separate research groups. The Breast Cancer Resistance Protein (BCRP) was obtained through cloning in a breast cancer lineage resistant to doxorubicin, an antibiotic with antitumor properties similar in structure to DNR [40]. The name ABC Placental Protein (ABCP) was given due to the high expression of this transporter in the human placenta [41]. And finally, Mitoxantrone Resistance Protein was named after the discovery of two genes expressed that conferred resistance to the anticancer drug mitoxantrone in several human cancer cell lines [42]. ABCG2 is a 655 amino acid, 72 kDa transporter and has a polypeptide chain of both an NBD and a TMD and is categorized as a half-size transporter [43]. It is assumed that, in order to carry out the transport of substances across membranes, it organizes itself either as homodimers [44] or homotetramers [45]. This transporter is expressed in the placenta, liver, kidney, intestines and in the blood-brain barrier, where it performs cellular detoxification functions, similar to the transporters mentioned above [32,43]. ABCG2 is capable of transporting a wide variety of both positively and negatively charged molecules, organic anions and sulfated conjugates [46].
Additionally, ABCG2 is highly expressed in human hematopoietic stem cells, being a marker of the side population phenotype, whose expression is reduced with the differentiation of these cells [47]. The role of ABCG2 in these cells is still unclear, but Krishnamurthy and colleagues demonstrated that ABCG2 binds heme and that, under hypoxic conditions, cells can upregulate ABCG2 expression via the Hypoxia-Inducible Factor transcription factor-1 (HIF-1), and use ABCG2 to reduce the accumulation of heme and porphyrins [48].
Metastasis and epithelial-mesenchymal transition
It is widely established that the Extracellular Matrix (ECM) is capable of regulating the development and homeostasis of organs and tissues, and that sustained alterations in ECM components can lead to the growth of neoplasms [49]. In solid tumors, some cells often acquire a more aggressive phenotype, gaining the ability to invade adjacent tissues and spread to distant organs or tissues, a phenomenon known as metastasis [50]. In carcinomas (malignant epithelial tumors), one of the first events associated with the induction of metastases is the Epithelial-Mesenchymal Transition (EMT). EMT is a process characterized basically by the loss of epithelial characteristics, such as tight junctions and polarized organization, and the gain of characteristics typical of connective tissue cells, giving the cells a greater degree of migration and invasiveness. During this process, the cells may show a capacity for self-renewal, similar to that seen in stem cells [51].
A large number of cellular processes are triggered by EMT, including: alteration of the expression pattern of specific surface proteins, reduction of epithelial cell-cell adhesion receptors E-cadherin and induction of typical mesenchymal proteins such as (N-cadherin, Vimentin, and Fibronectin [52-54], also cytoskeleton rearrangement with a concomitant loss of cell polarity [55], re-modeling of the extracellular matrix through increased expression of digestive enzymes [56-58], and changes in the expression of specific microRNAs (miRNAs) [59-61], which may target transcription factors involved in triggering the process, as well as proteins that play a key role in the transition, such as E-cadherin. All these aforementioned events occur from the activation of a set of transcription factors specific to the EMT program (EMT-TFs), including zinc-finger E-box-binding homeobox (Zeb) 1 and 2, Snail, (also known as SNAI1), Slug (also known as SNAI2) and Twist-related protein 1 (Twist-1) [62,63].
In cancer, EMT activation does not promote a complete differentiation into a mesenchymal state, instead consisting on a partial/hybrid process crucial for tumor progression that induces a heterogeneous cellular transdifferentiation, leading to the onset of several tumor subpopulations, which may differ in their (in) differentiation status, migration, invasiveness and induction of metastasis [64,65]. Moreover, there is a remarkable role of the tumor microenvironment (with different niches) in determining the extent of EMT in tumor cells, in which subpopulations that show more mesenchymal characteristics are those usually located near to the endothelium and inflammatory cells of the immune system [64].
Among the endogenous agents that induce EMT, TGF-β is one of the most studied, as this cytokine is actively produced by tumors during their development [66] and has been described as a potent inducer of this process in various cell types in studies both in vitro [67-69] and in vivo [70-72]. The cellular pathways triggered by TGF-β-induced EMT involve the canonical activation, mediated by Smad proteins, which promotes the activation of the EMT-TFs Snail, slug, Twist-1 and Zeb [73] and non-canonical activation, leading to the activation of several other pathways, such as PI3K/Akt, Rho/ROCK, as well as ERK, p38 and JNK, which act orchestrating the regulation of several processes, such as proliferation, differentiation, reorganization of the cytoskeleton and apoptosis [74-77]. In addition, TGF-β action during EMT also occurs by means of cooperation with other pathways, such as Wnt (through the activation of β-catenin) [78-80] and Notch pathway via the formation of a positive regulatory loop, in which activation of the Notch pathway leads to further activation of SMADs (from canonical TGF-β-mediated EMT), as well as increased production of TGF-β and its receptors. Moreover, TGF-β -induced EMT induces a huge cellular metabolic reprogramming, by means of increased glucose uptake due to enhanced expression of GLUT1 transporter [81] and also promotes a shift in cell energy metabolism, by favoring glycolysis through direct activation of hexokinases, phosphofructokinases and pyruvate kinases [82].
Noteworthy, it has also been shown that modifications in ECM components themselves could be related to the induction of EMT. In this context, it has been reported that the production of an aberrant fibronectin, containing an O-glycosylation in its variable chain, was associated with the induction of EMT in both tumor cells and in epithelial cells with a normal phenotype. Since this aberrant fibronectin had a similar structure to fibronectin isolated from embryos or transformed cells, it was named oncofetal fibronectin. Furthermore, the simple addition of oncofetal fibronectin to carcinoma cell lines was able to induce EMT, a fact that did not occur when fibronectin lacking such O-glycosylation in the variable chain was added to the same cultures [83-85]. More recently, it has also been demonstrated that multiresistant MCF-7 lines express high levels of oncofetal fibronectin and the knockdown of pp-GalNAc-T6 (enzyme involved in glycoprotein biosynthesis) partially reversed the MDR phenotype [86].
Tumor progression: The onset of metastasis and its relation with chemotherapy resistance
Whilst cancer usually originates from alterations occurring on a single tissue, it is composed by a cellular population that presents marked heterogeneity. In this context, only a small populapopulation is capable of originating metastases, cells thus called Metastasis Initiating Cells (MIC) [9]. MIC show great plasticity, being able to undergo a transdifferentiation process that culminates in their departure from the original tumor, survival as circulating tumor cells and the later homing into a secondary metastatic niche.
The establishment of metastatic cells is a phenomenon characterized by cell cycle arrest that drives a state of dormancy or quiescence, which is vital for evading recognition by the immune system [87]. On the whole, metastatic cells in circulation settle in the metastatic tissue nearby capillaries, remaining in a quiescent state due to mediators such as thrombospondin-1 or TGF-β [88,89]. This fact explains why often after surgical removal of the primary tumor, patients do not show clinical signs of the disease for many months, but further exhibit an aggressive growth of the tumor in metastatic sites.
However, the molecular events involved in this process still remain partially known. An example is the fact that primary tumors are able to prioritize distant organs and tissues in order to create a favorable niche for colonization by metastatic cells. Such event is possible because primary tumors release several factors including hormones, cytokines and chemokines in a soluble form or via extracellular vesicles that prime those organs and tissues to support the newly arrived metastatic cells, thus creating a so called pre-metastatic niche. Such priming induces the chemoattraction of bone marrow progenitors to the pre-metastatic niche, where they will act by creating a favorable environment to metastatic cells, that will increase their survival capacity and also promote resistance to chemotherapy drugs [90-92]. The onset of chemoresistance seems to occur as a result of multiple gene expression profiles within particular tumor cells, thus originating specific metastatic subpopulations that may exhibit either one or several resistance mechanisms simultaneously [93-95].
Aiming to understand possible alterations in gene patterns during tumor progression, the expression profile of 380 genes related to the induction of resistance to treatment was evaluated in patients with acute myeloid leukemia at the diagnosis of the disease and compared with gene expression patterns at relapse. Among the genes observed, those showing the greatest increase were genes encoding Glutathione S transferase family members (proteins that mediate drug detoxification), bcl-2 (known for its anti-apoptotic activity) and transporters from the ABC superfamily, such as ABCB1, ABCC3 and ABCG2 [96].
Another example of relevant cancer inducer genes is Ras protooncogene. How Ras proteins are connected to cancer progression is long known, as this GTPase is at the core of several cell signaling pathways related to response to tyrosine kinase receptors, G-protein coupled receptors and integrins. When mutated, and consequently remaining on its active state even in the absence of receptor stimulation, cells undergo proliferation, apoptosis inhibition, metabolic regulation and increased cellular motility and invasion [97]. Although it is a consensus that members of the Ras family of proteins are often mutated in cancer, there is a significant disparity in the frequency of mutations when evaluating different tumor types.
Nonetheless, despite this discrepancy, nearly 20% of all types of tumors exhibit Ras mutations, corroborating the relevance of Ras-signaling pathways for disease progression [98]. Moreover, Ras mutations may also concur to the onset of drug resistance, as demonstrated in a multicenter study, in which patients with metastatic colorectal cancer presenting Ras mutations developed resistance against cetuximab chemotherapy, resulting in roughly 50% decrease in progression-free survival when compared to patients with wild-type Ras [99]. Albeit resistance to chemotherapy is a multifactorial phenotype, there is large evidence that hyperactivation of Ras-signaling pathways, such as ERK1/2 or the PI3K/AkT pathway, may account for the increase of ABC-related resistance, via the induction of ABCB1, ABCC1 and ABCG2 [100-104].
In vitro experiments regarding drug-induced EMT using adriamycin demonstrated the upregulation of ABCB1 following EMT triggering. Cells undergoing EMT, identified by Twist-1 upregulation, also exhibited an increase in ABCB1 expression, though the later was also linked to a significant induction of cell death [105]. Drug efflux was the first mechanism described nearly 50 years ago and is the most studied and characterized mechanism by which cancer cells may acquire the multidrug resistance phenotype. Its importance may be demonstrated by evaluating the plethora of chemotherapy drugs which are substrates of one (or more) ABC transporters, like anthracyclines, alkylating agents, vinca alkaloids and tyrosine kinase inhibitors, among others.
By utilizing more modern detection techniques, such as RNAseq, it has been observed that various types of tumors express high levels of ABC proteins, which can vary by over a thousand times between different patients. Particularly, when evaluating the expression of the ABCB1 transporter, high expression was noted in 30 different types of tumors, remarkably on pancreatic tumors, glioblastomas, renal carcinomas, and diffuse large B-cell lymphomas. As for ABCG2, whose expression has also been described in various types of tumors, the highest expression levels were detected in prostate tumors, thyroid tumors, cholangiocarcinoma, glioma, and glioblastoma [106]. Moreover, in a comprehensive analysis of ABC transporters in pancreatic cancer, it was discovered that several ABC proteins, like ABCB4, ABCB11, ABCC1, ABCC3, ABCC5, ABCC10, and ABCG2 exhibited a notable increase in mRNA expression within macrodissected tumors, when compared to healthy tissues [107]. Our group also demonstrated that cisplatin-resistant adenocarcinoma cells exhibit EMT induction, presenting also enhanced migration when compared to parental cells, along with increased expression ABCB1, ABCC1, and ABCG2 [108].
Despite this, it is noteworthy that the overwhelming majority of studies evaluating the relationship between the function of ABC proteins and tumor progression focused on the role of chemotherapy drugs. Furthermore, many of the studies did not evaluate the relationship between a possible modulation of ABC proteins expression and an increase in the protein transport capacity or resistance capacity of the tumor itself. Trying to unveil this matter, our group sought to investigate the action of TGF-β-induced EMT on the expression and activity of ABCB1 and ABCC1. Corroborating previous findings, we have observed that TGF-β-induced EMT promoted a slight rise in ABCB1 expression (with no change in protein activity), in a small portion of the A549 cell population that correlated with the induction of EMT. Interestingly, ABCB1 inhibition did not promote any changes in TGF-β-induced EMT, though persistent ABCB1 inhibition was correlated with increase of the mesenchymal markers Fibronectin and Snail. On the other hand, physiological EMT resulted in a significant upregulation of both ABCC1 expression and activity. Moreover, ABCC1 inhibition on the course of TGF-β-induced EMT partially reverted Snail induction [109]. Since ABCC1 activity is involved in controlling intracellular redox status by extruding reduced or oxidized GSH [110,111], ABCC1 impairment may have hindered the full progression of EMT in A549 cells.
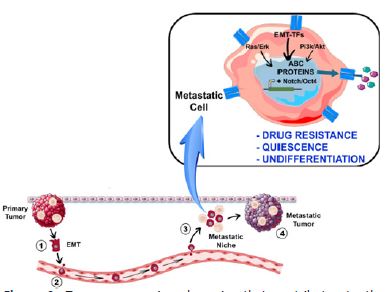
A question that still remains is how cancer progression and chemoresistance induction are indeed connected. Numerous studies provide substantial evidence supporting the idea that induction of metastasis would confer a stem cell-like phenotype to tumor cells, ultimately giving rise to Cancer Stem Cells (CSCs). In this regard, the activation of EMT would trigger the expression of specific genes associated with an undifferentiated stem-like state, including Oct4 and Notch. This process leads to the onset of a Side Population (SP) phenotype, characterized by overexpression of ABC transporters such as ABCB1 and ABCG2, enabling the efficient transport of various toxic substrates, such as xenobiotics and chemotherapeutic drugs, as summarized in Figure 2 [112-115].
The determination of the stem cell-like state relies on the dynamic EMT-regulatory network. This is exemplified by the cooperative actions of transcription factors Slug and Sox9, which are responsible for establishing the CSC state and influencing the tumorigenic and metastasis-seeding abilities of human breast cancer [116]. Additionally, Zeb-1, a master inducer of EMT, has been identified as a key regulator of stemness status, tumorigenicity, and cell plasticity in pancreatic cancer [117]. Regarding TGF-β-induced EMT, both Zeb2 and Twist1 were highly induced during EMT and mutual inhibition of these transcription factors significantly reduced metastasis in a mice model of colorectal carcinoma [118].
SP cells were identified by separation of hematopoietic cells based on their location in the fluorescence-activated cell sorting scatter plot, specifically in the Hoechst Blue-low and Hoechst Red-low quadrant, distinct from the majority of cells [119,120]. However, the so-called SP population is not entirely uniform. Detection of SP in different tissues has unveiled a remarkable heterogeneity, typically show casing an undifferentiated nature, albeit not consistently accompanied by the presence of characteristic stem cell markers, thus belonging to distinct stages of differentiation [121-123]. Within this context, it is inadequate to solely rely on the expression of ABC transporters, like ABCG2 (which is highly expressed in stem cells), to identify stem cells. This was evident when cells are purified and isolated from the entire bone marrow population solely based on ABCG2 expression, since ABCG2-positive population showed a reduced capacity to form colonies, a common feature of stem cells [124]. Furthermore, functional as says have shown that ABCG2 knockout mice have normal numbers of hematopoietic stem cells in bone marrow, though with the loss of this membrane transporter they were located outside the SP region, thus demonstrating that the terms side population and stem cells should not be employed as synonyms [125]
Further investigations have provided additional support for the heterogeneity of SP populations and its clinical outcome even in hematological tumors. In Acute Myeloid Leukemia (AML) blasts from human patients with mesenchymal stromal cells derived from bone marrow, it was revealed an elevated activity of ABCB1, ABCG2, and ABCC1, leading to the emergence of an SP phenotype [126]. Another study demonstrated that increase in expression and function of those ABC transporters in co-expression was shown to correlate with increased expression of early progenitor markers, such as c-kit, and with poor prognosis of patients with AML, especially due to poor response to chemotherapy [127].
Conclusion
Cancer is recognized as a major global public health challenge, ranking first in the top 10 leading causes of death worldwide [128]. Concerning cancer treatment, both the development of metastasis and the emergence of the multidrug resistance phenotype pose as challenges to cancer chemotherapy, where overexpression of ABC superfamily members is highlighted as a negative prognosis for the disease, significantly reducing the survival rate of cancer patients [12,129-133].
In this particular context, it is crucial to understand the mechanisms associated with these complex events in order to pursue more effective therapies against cancer. Unfortunately, despite the robust in vivo correlation between the overexpression of ABC transporters and a poor prognosis in cancer, along with the description of a plethora of molecules capable of inhibiting the activity of these pumps, most therapies based on the combination of pharmacological ABC inhibitors with chemotherapeutic agents have not revealed the expected beneficial effect.
Such observation is grounded in numerous studies that have demonstrated either no positive impact or the induction of toxicity in various tissues [134-136]. Moreover, such association has resulted in other side effects, including changes in the metabolism of various compounds and the facilitation of drug-drug interactions [137]. Finally, it was observed that certain inhibition conditions led to a significant increase in the transport activity of ABC proteins, which completely contradicts the rationale for using ABC inhibitors as adjuvants to therapy [138]. Although the use of pharmacological inhibitors has shown great effectiveness in eliminating resistant tumors in vitro [139,140], the aforementioned adverse outcomes in vivo were a result of physiological expression of ABC proteins in different organs and tissues [15,135].
Therefore, the search for therapies capable of evading the transport of a broad spectrum of chemotherapeutic substrates, mediated by ABC transporters, is of utmost importance. Understanding the complex mechanisms that occur during tumor progression, initiated by pathways leading to EMT triggering and the acquisition of an undifferentiated phenotype, is essential. These processes collectively contribute to the development of a broad-spectrum resistance phenotype to chemotherapeutics. In that way, it will be possible to obtain new therapeutic targets for resistant tumors or, alternatively, bring light to the conception of new approaches that could take advantage of the overexpression of ABC proteins against the tumor itself; for instance, by improving methods for the safe and effective use of collateral sensitivity as a tumor’s Achilles’ heel.
Abbreviations: ABC: ATP-Binding Cassette; ABCB1: Subfamily B Member 1; ABCB11: subfamily B Member 11; ABCB4: Subfamily B, Member 4; ABCC1: Subfamily C Member 1; ABCC10: Subfamily C Member 10; ABCC3: Subfamily C Member 3; ABCC5: Subfamily C Member 5; ABCG2: Subfamily G Member 2; AML: Acute Myeloid Leukemia; ASC: Adipose-Derived Stromal Cells; BCRP: Breast Cancer Resistance Protein; CSC: Cancer Stem Cells; DNR: Daunorubicin; ECM: Extracellular Matrix; EMT: Epithelial-Mesenchymal Transition; EMT-TFs: Epithelial Mesenchymal Transition Transcription Factors; GLUT1: Glucose Transporter 1; GSH: Reduced Glutathione; HIF-1: Hypoxia-Inducible Factor Transcription Factor-1; MDR: Multi-Drug Resistance; MDR1: multidrug Resistance Protein-1; MIC: Metastasis Initiating Cells; NBD: Nucleotide Binding Domain; NSC-32946: Methylglyoxal bisguanylhydrazone, di-hydrochloride; NSC-38280: 2-chloro-4’, 4»- di (2-imidazolin-2-yl) terephthalanilide dihydrochloride; SP: Side Population; TGF-β: Transforming Growth Factor Beta; TMD: Transmembrane Domain; Twist-1: Twist-Related Protein 1; Zeb: Zinc-finger E-box-binding homeobox.
Declarations
Author contributions: Conceptualization: K.M.d.C., R.d.C.V., L.F.-d.-L, L.M.F, J.O.P. and L.M.-P; writing—original draft preparation: K.M.d.C. and R.d.C.V.; writing—review and editing: K.M.d.C., R.d.C.V., L.F.-d.-L., L.M.F., J.O.P. and L.M.-P; supervision: R.d.C.V., J.O.P. and L.M.-P; funding acquisition, J.O.P. and L.M.-P. All authors have read and agreed to the published version of the manuscript.
Funding: This research was funded by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnolόgico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
References
- HS Ritchie, F Roser M. Causes of death. 2023.
- MR Roser, H. Cancer. 2023.
- N. National Cancer Institute. «What Is Cancer?,»
- CR UK. Types of cancer. https://www.cancerresearchuk.org/about-cancer/what-is-cancer/how-cancer-starts/types-of-cancer.
- D Hanahan, RA Weinberg. The hallmarks of cancer. Cell. 2000; 100: 57-70.
- D Hanahan, RA Weinberg. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-74.
- D Hanahan. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022; 12: 31-46.
- J Fares, MY Fares, HH Khachfe, et al. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther. 2020; 5: 28.
- K Ganesh, J Massague. Targeting metastatic cancer. Nat Med. 2021; 27: 34-44.
- V Mittal. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol. 2018; 13: 395-412.
- G Sannino, A Marchetto, T Kirchner, Grunewald TGP. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transition in Mesenchymal Tumors: A Paradox in Sarcomas?. Cancer Res. 2017; 77: 4556-4561.
- PS Steeg. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006; 12: 895-904.
- S Norouzi, M Gorgi Valokala, F Mosaffa, Zirak MR, Zamani P, et al. Crosstalk in cancer resistance and metastasis. Crit Rev Oncol Hematol. 2018; 132: 145-153.
- W Muriithi, LW Macharia, CP Heming, Echevarria JL, Nyachieo A, et al. ABC transporters and the hallmarks of cancer: roles in cancer aggressiveness beyond multidrug resistance. Cancer Biol Med. 2020; 17: 253-269.
- M Dean, K Moitra, R Allikmets. The human ATP-binding cassette (ABC) transporter superfamily. Hum Mutat. 2022; 43: 1162-1182.
- DJ Hutchison. Cross Resistance and Collateral Sensitivity Studies in Cancer Chemotherapy. Adv Cancer Res. 1963; 7: 235-50.
- FRK HEILMAN EC. The influence of 11-dehydro-17hydroxycorticosterone (compound E) on the growth of a malignant tumor in the mouse. Endocrinology. 1944; 34: 416-420.
- JH Burchenal, E Robinson, et al. The induction of resistance to 4-amino-N10-methylpteroylglutamic acid in a strain of transmitted mouse leukemia. Science. 1950; 111: 2875.
- JH Burchenal, SF Johnston, GB Waring. Mechanisms of amethopterin resistance in leukemia. I. Effects of weak folic acid antagonists on mouse leukemias. Proc Soc Exp Biol Med. 1951; 78: 348-51.
- JH Burchenal, LF Webber, SF Johnston. Mechanisms of amethopterin resistance in leukemia. II. Effect of cortisone on sensitive and resistant mouse leukemias. Proc Soc Exp Biol Med. 1951; 78: 352-4.
- GA Fischer. Studies of the culture of leukemic cells in vitro. Ann N Y Acad Sci. 1958; 76: 673-80.
- RH Adamson, VT Oliverio, C Denham, et al. Concentration of methylglyoxal-bis-guanylhydrazone-C14 in mouse leukemia L1210 and a resistant variant. Life Sci. 1962; 7: 493-6.
- A Sivak, WI Rogers, I Wodinsky, et al. Distribution and Effects of 4’, 4»-Di(2-Imidazolin-2-Yl) Terephthalanilide Dihydrochloride Hemihydrate and Its 2-Chloro Analogue in Lymphocytic Leukemias L1210 and P388. J Natl Cancer Inst. 1964; 33: 457-65.
- WI Rogers, A Sivak, CJ Kensler. Physiologic Disposition Studies of Some Substituted Phthalanilides. Cancer Chemother Rep. 1964; 38: 9-16.
- DW Yesair, FA Kohner, WI Rogers, et al. Relationship of phthalanilide-lipid complexes to uptake and retention of 2-chloro-4’,4»-di(2-imidazolin-2-yl)terephthalanilide (NSC 60339) by sensitive and resistant P388 leukemia cells. Cancer Res. 1966; 26: 202-7.
- D Kessel, V Botterill, I Wodinsky. Uptake and retention of daunomycin by mouse leukemic cells as factors in drug response. Cancer Res. 1968; 28: 938-41.
- K Dano. Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells. Biochim Biophys Acta. 1973; 323: 466-83.
- RL Juliano, V Ling. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976; 455: 152-62.
- C Pilotto Heming, W Muriithi, L Wanjiku Macharia, et al. P-glycoprotein and cancer: what do we currently know?. Heliyon. 2022; 8: e11171.
- DR Bell, JH Gerlach, N Kartner, et al. Detection of P-glycoprotein in ovarian cancer: a molecular marker associated with multidrug resistance. J Clin Oncol. 1985; 3: 311-5.
- F Thiebaut, T Tsuruo, H Hamada, et al. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA. 1987; 84: 7735-8.
- EM Leslie, RG Deeley, SP Cole. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005; 204: 216-37.
- FJ Sharom. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008; 9: 105-27.
- R Silva, V Vilas-Boas, H Carmo, et al. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. 2015; 149: 1-123.
- SP Cole, G Bhardwaj, JH Gerlach, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992; 258: 1650-4.
- L Couture, JA Nash, J Turgeon. The ATP-binding cassette transporters and their implication in drug disposition: a special look at the heart. Pharmacol Rev. 2006; 58: 244-58.
- AJ Slot, SV Molinski, SP Cole. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011; 50: 179-207.
- SP Cole. Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol. 2014; 54: 95-117.
- MJ Flens, GJ Zaman, P van der Valk, et al. Tissue distribution of the multidrug resistance protein. Am J Pathol. 1996; 148: 1237-47.
- LA Doyle, W Yang, LV Abruzzo, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998; 95: 15665-70.
- R Allikmets, LM Schriml, A Hutchinson, et al. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998; 58: 5337-9.
- K Miyake, L Mickley, T Litman, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999; 59: 8-13.
- F Staud, P Pavek. Breast cancer resistance protein (BCRP/ABCG2). Int J Biochem Cell Biol. 2005; 37: 720-5.
- A Bhatia, HJ Schafer, CA Hrycyna. Oligomerization of the human ABC transporter ABCG2: evaluation of the native protein and chimeric dimers. Biochemistry. 2005; 44: 10893-904.
- K Wong, SJ Briddon, ND Holliday, et al. Plasma membrane dynamics and tetrameric organisation of ABCG2 transporters in mammalian cells revealed by single particle imaging techniques. Biochim Biophys Acta. 2016; 1863: 19-29.
- D Pena-Solorzano, SA Stark, B Konig, et al. ABCG2/BCRP: Specific and Nonspecific Modulators. Med Res Rev. 2017; 37: 987-1050.
- S Zhou, JD Schuetz, KD Bunting, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a momolecular determinant of the side-population phenotype. Nat Med. 2001; 7: 1028-34.
- P Krishnamurthy, DD Ross, T Nakanishi, et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004; 279: 24218-25.
- MW Pickup, JK Mouw, VM Weaver. The extracellular matrix modulates the hallmarks of cancer, EMBO Rep. 2014; 15: 1243-53.
- S Valastyan, RA Weinberg, Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147: 275-92.
- SA Mani, W Guo, MJ Liao, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133: 704-15.
- M Wang, D Ren, W Guo, et al. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. Int J Oncol. 2016; 48: 595-606.
- CY Liu, HH Lin, MJ Tang, et al. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget. 2015; 6: 15966-83.
- TC Lin, CH Yang, LH Cheng, et al. Fibronectin in Cancer: Friend or Foe. Cells. 2019; 9: 1.
- A Datta, S Deng, V Gopal, et al. Cytoskeletal Dynamics in Epithelial-Mesenchymal Transition: Insights into Therapeutic Targets for Cancer Metastasis. Cancers (Basel). 2021; 13: 8.
- MD Sternlicht, A Lochter, CJ Sympson, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999; 98: 137-46.
- MD Martin, KJ Carter, SR Jean-Philippe, et al. Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 2008; 68: 6251-9.
- J Winkler, A Abisoye-Ogunniyan, KJ Metcalf, et al. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020; 11: 5120.
- PA Gregory, AG Bert, EL Paterson, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008; 10: 593-601.
- L Ma, J Young, H Prabhala, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010; 12: 247-56.
- R Kumarswamy, G Mudduluru, P Ceppi, et al. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012; 130: 2044-53.
- R Kalluri, RA Weinberg. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119: 1420-8.
- MP Stemmler, RL Eccles, S Brabletz, et al. Non-redundant functions of EMT transcription factors. Nat Cell Biol. 2019; 21: 102-112.
- Pastushenko, A Brisebarre, A Sifrim, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018; 556: 463-468.
- C Kroger, A Afeyan, J Mraz, et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci USA. 2019; 116: 7353-7362.
- CB Trelford, L Dagnino, GM Di Guglielmo. Transforming growth factor-beta in tumour development. Front Mol Biosci. 2022; 9: 991612.
- A Dhasarathy, D Phadke, D Mav, et al. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One. 2011; 6: e26514.
- V Maturi, S Enroth, CH Heldin, et al. Genome-wide binding of transcription factor ZEB1 in triple-negative breast cancer cells. J Cell Physiol. 2018; 233: 7113-7127.
- SA Mikheeva, AM Mikheev, A Petit, et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol Cancer. 2010; 9: 194.
- G Portella, SA Cumming, J Liddell, et al. Transforming growth factor beta is essential for spindle cell conversion of mouse skin carcinoma in vivo: implications for tumor invasion. Cell Growth Differ. 1998; 9: 393-404.
- M Oft, KH Heider, H Beug. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998; 8: 1243-52.
- A Bruna, RS Darken, F Rojo, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007; 11: 147-60.
- Xu, S Lamouille, R Derynck. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009; 19: 156-72.
- Q Wang, X Yang, Y Xu, et al. RhoA/Rho-kinase triggers epithelial-mesenchymal transition in mesothelial cells and contributes to the pathogenesis of dialysis-related peritoneal fibrosis, Oncotarget. 2018; 9: 14397-14412.
- M Navandar, A Garding, SK Sahu, et al. ERK signalling modulates epigenome to drive epithelial to mesenchymal transition, Oncotarget. 2017; 8: 29269-29281.
- A Rodriguez-Garcia, P Samso, P Fontova, et al. TGF-beta1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017; 284: 3437-3454.
- Y Hao, D Baker, P Ten Dijke. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci. 2019; 20: 11.
- E Dzialo, M Czepiel, K Tkacz, et al. WNT/beta-Catenin Signaling Promotes TGF-beta-Mediated Activation of Human Cardiac Fibroblasts by Enhancing IL-11 Production. Int J Mol Sci. 2021; 22: 18.
- Y Yoo, BJ Ku, TH Kim, et al. beta-catenin activates TGF-beta-induced epithelial-mesenchymal transition in adenomyosis. Exp Mol Med. 2020; 52: 1754-1765.
- Xu, WH Cui, WC Zhou, et al. Activation of Wnt/beta-catenin signalling is required for TGF-beta/Smad2/3 signalling during myofibroblast proliferation. J Cell Mol Med. 2017; 21: 1545-1554.
- W Li, Z Wei, Y Liu, et al. Increased 18F-FDG uptake and expression of Glut1 in the EMT transformed breast cancer cells induced by TGF-beta. Neoplasma. 2010; 57: 234-40.
- W Hua, P Ten Dijke, S Kostidis, et al. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci. 2020; 77: 2103-2123.
- Freire-de-Lima, K Gelfenbeyn, Y Ding, et al. Involvement of O-glyglycosylation defining oncofetal fibronectin in epithelial-mesenchymal transition process, Proc Natl Acad Sci USA, 2011; 108: 17690-5.
- Y Ding, K Gelfenbeyn, L Freire-de-Lima, et al. Induction of epithelial-mesenchymal transition with O-glycosylated oncofetal fibronectin. FEBS Lett. 2012; 586: 1813-20.
- F Alisson-Silva, L Freire-de-Lima, JL Donadio, et al. Increase of O-glycosylated oncofetal fibronectin in high glucose-induced epithelial-mesenchymal transition of cultured human epithelial cells. PLoS One. 2013; 8: e60471.
- JSD Reis, M Santos, KM da Costa, et al. Increased expression of the pathological O-glycosylated form of oncofetal fibronectin in the multidrug resistance phenotype of cancer cells. Matrix Biol. 2023; 118: 47-68.
- S Malladi, DG Macalinao, X Jin, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell. 2016; 165: 45-60.
- CM Ghajar, H Peinado, H Mori, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013; 15: 807-17.
- P Bragado, Y Estrada, F Parikh, et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol. 2013; 15: 1351-61.
- RN Kaplan, RD Riba, S Zacharoulis, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche Nature. 2005; 438: 820-7.
- S Acharyya, T Oskarsson, S Vanharanta, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012; 150: 165-78.
- H Peinado, H Zhang, IR Matei, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017; 17: 302-317.
- RS Kerbel, H Kobayashi, CH Graham. Intrinsic or acquired drug resistance and metastasis: are they linked phenotypes?. J Cell Biochem. 1994; 56: 37-47.
- RG Amado, M Wolf, M Peeters, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008; 26: 1626-34.
- CA Stewart, CM Gay, Y Xi, et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat Cancer. 2020; 1: 423-436.
- C Patel, L Stenke, S Varma, et al. Multidrug resistance in relapsed acute myeloid leukemia: evidence of biological heterogeneity. Cancer. 2013; 119: 3076-83.
- RC Gimple, X Wang. RAS: Striking at the Core of the Oncogenic Circuitry, Front Oncol. 2019; 9: 965.
- A Prior, FE Hood, JL Hartley. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020; 80: 2969-2974.
- HL Tsai, CC Lin, YC Sung, et al. The emergence of RAS mutations in patients with RAS wild-type mCRC receiving cetuximab as first-line treatment: a noninterventional, uncontrolled multicenter study. Br J Cancer. 2023; 129: 947-955.
- H Xu, Y Li, L Chen, et al. SIRT2 mediates multidrug resistance in acute myelogenous leukemia cells via ERK1/2 signaling pathway. Int J Oncol. 2016; 48: 613-23.
- PL Tazzari, A Cappellini, F Ricci, et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia. 2007; 21: 427-38.
- R Chen, XH Jia, H Wang, et al. Timosaponin A-III reverses multi-drug resistance in human chronic myelogenous leukemia K562/ADM cells via downregulation of MDR1 and MRP1 expression by inhibiting PI3K/Akt signaling pathway. Int J Oncol. 2016; 48: 2063-70.
- H Tomiyasu, M Watanabe, K Sugita, et al. Regulations of ABCB1 and ABCG2 expression through MAPK pathways in acute lymphoblastic leukemia cell lines. Anticancer Res. 2013; 33: 5317-23.
- G Dharmapuri, R Doneti, GH Philip, et al. Celecoxib sensitizes imatinib-resistant K562 cells to imatinib by inhibiting MRP1-5, ABCA2 and ABCG2 transporters via Wnt and Ras signaling pathways. Leuk Res. 2015; 39: 696-701.
- QQ Li, JD Xu, WJ Wang, et al. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009; 15: 2657-65.
- RW Robey, KM Pluchino, MD Hall, et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018; 18: 452-464.
- B Mohelnikova-Duchonova, V Brynychova, M Oliverius, et al. Differences in transcript levels of ABC transporters between pancreatic adenocarcinoma and nonneoplastic tissues. Pancreas. 2013; 42: 707-16.
- M da Fonseca, VA da Silva, KM da Costa, et al. Resistance to cisplatin in human lung adenocarcinoma cells: effects on the glycophenotype and epithelial to mesenchymal transition markers. Glycoconj J. 2022; 39: 247-259.
- M da Costa, L Freire-de-Lima, LM da Fonseca, et al. ABCB1 and ABCC1 Function during TGF-beta-Induced Epithelial-Mesenchymal Transition: Relationship between Multidrug Resistance and Tumor Progression. Int J Mol Sci. 2023; 24: 7.
- CF Mueller, JD Widder, JS McNally, et al. The role of the multidrug resistance protein-1 in modulation of endothelial cell oxidative stress. Circ Res. 2005; 97: 637-44.
- SY Lee, MK Ju, HM Jeon, et al. Reactive oxygen species induce epithelial‑mesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx‑2/Snail signaling pathways in MCF‑7 cells. Mol Med Rep. 2019; 20: 2339-2346.
- D Wang, P Lu, H Zhang, et al. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients, Oncotarget. 2014; 5: 10803-15.
- Liu H, Zhu Y, Liao, et al. Inhibition of Wnt/beta-catenin pathway reverses multi-drug resistance and EMT in Oct4(+)/Nanog(+) NSCLC cells. Biomed Pharmacother. 2020; 127: 110225.
- V Gustafsson, X Zheng, T Pereira, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005; 9: 617-28.
- A Mohan, RR Raj, G Mohan, et al. Reporters of Cancer Stem Cells as a Tool for Drug Discovery. Front Oncol. 2021; 11: 669250.
- W Guo, Z Keckesova, JL Donaher, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012; 148: 1015-28.
- AM Krebs, J Mitschke, M Lasierra Losada, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017; 19: 518-529.
- Y Zheng, M Dai, Y Dong, et al. ZEB2/TWIST1/PRMT5/NuRD Multicomplex Contributes to the Epigenetic Regulation of EMT and Metastasis in Colorectal Carcinoma. Cancers (Basel). 2022; 14: 14.
- A. Goodell, Stem cell identification and sorting using the Hoechst 33342 side population (SP), Curr Protoc Cytom, vol. Chapter 9, pp. Unit9 18, Nov, 2005.
- A Mayle, M Luo, M Jeong, et al. Flow cytometry analysis of murine hematopoietic stem cells. Cytometry A. 2013; 83: 27-37.
- DC Andersen, HD Schroder, CH Jensen. Non-cultured adipose-derived CD45- side population cells are enriched for progenitors that give rise to myofibres in vivo. Exp Cell Res. 2008; 314: 2951-64.
- H Masuda, T Maruyama, CE Gargett, et al. Endometrial side population cells: potential adult stem/progenitor cells in endometrium. Biol Reprod. 2015; 93: 84.
- GA Challen, MH Little. A side order of stem cells: the SP phenotype. Stem Cells. 2006; 24: 3-12.
- CS Naylor, E Jaworska, K Branson, et al. Side population/ABCG2-positive cells represent a heterogeneous group of haemopoietic cells: implications for the use of adult stem cells in transplantation and plasticity protocols, Bone Marrow Transplant. 2005; 35: 353-60.
- S Zhou, JJ Morris, Y Barnes, et al. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci USA. 2002; 99: 12339-44.
- L Boutin, P Arnautou, A Trignol, et al. Mesenchymal stromal cells confer chemoresistance to myeloid leukemia blasts through Side Population functionality and ABC transporter activation. Haematologica. 2020; 105: 987-9998.
- B Liu, LJ Li, X Gong, et al. Co-expression of ATP binding cassette transporters is associated with poor prognosis in acute myeloid leukemia. Oncol Lett. 2018; 15: 6671-6677.
- S Dattani, Spooner F, Ritchie H, Roser M. Causes of Death. Our World in Data. 2023.
- F Zou, M Seike, R Noro, et al. Prognostic significance of ABCB1 in stage I lung adenocarcinoma. Oncol Lett. 2017; 14: 313-321.
- DH Sabnis, LCD Storer, JF Liu, et al. A role for ABCB1 in prognosis, invasion and drug resistance in ependymoma. Sci Rep. 2019; 9: 10290.
- X Tong, J Zhao, Y Zhang, et al. Expression levels of MRP1, GST-pi, and GSK3beta in ovarian cancer and the relationship with drug resistance and prognosis of patients. Oncol Lett. 2019; 18: 22-28.
- Q Dong, C Zhou, H Ren, et al. Lactate-induced MRP1 expression contributes to metabolism-based etoposide resistance in non-small cell lung cancer cells, Cell Commun Signal. 2020; 18: 167.
- A Tivnan, Z Zakaria, C O’Leary, et al. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front Neurosci. 2015; 9: 218.Cancer. Clin Pharmacol Ther. 2020; 108: 671-680.
- TB Stage, C Mortensen, S Khalaf, et al. P-Glycoprotein Inhibition Exacerbates Paclitaxel Neurotoxicity in Neurons and Patients With Cancer. Clin Pharmacol Ther. 2020; 108: 671-680.
- G Szakacs, JK Paterson, JA Ludwig, et al. Targeting multidrug resistance in cancer, Nat Rev Drug Discov. 2006; 5: 219-34.
- S Daenen, B van der Holt, GE Verhoef, et al. Addition of cyclosporin A to the combination of mitoxantrone and etoposide to overcome resistance to chemotherapy in refractory or relapsing acute myeloid leukaemia: a randomised phase II trial from HOVON, the Dutch-Belgian Haemato-Oncology Working Group for adults. Leuk Res. 2004; 28: 1057-67.
- W Klinkhammer, H Muller, C Globisch, et al. Synthesis and biological evaluation of a small molecule library of 3rd generation multidrug resistance modulators. Bioorg Med Chem. 2009; 17: 2524-35.
- HS Kim, YD Min, CH Choi. Double-edged sword of chemosensitizer: increase of multidrug resistance protein (MRP) in leukemic cells by an MRP inhibitor probenecid. Biochem Biophys Res Commun. 2001; 283: 64-71.
- R Callaghan, F Luk, M Bebawy. Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy?. Drug Metab Dispos. 2014; 42: 623-31.
- RW Robey, S Shukla, EM Finley, et al. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1((R)). Biochem Pharmacol. 2008; 75: 1302-12.