Introduction
Most prostate cancers depend on androgens for their survival, growth, and progression. The androgens activate Androgen Receptors (AR) within the tumor cells, causing cell proliferation and tumor progression. Androgen receptors are also present within the normal prostate’s glandular epithelial cells and stromal cells and have a significant role in organ development and maintenance of normal prostatic function [1]. Charles Huggins and Clarence V. Hodges (1941) argued that prostate carcinomas are highly dependent on the presence of androgenic hormones for their growth and progression. Through their experiments, they showed that surgical removal of testes in patients with prostate cancer induces deprivation of androgen that may cause a marked regression of cancer [2]. Men with advanced prostate cancer are now routinely treated by androgen deprivation therapy. In addition to surgical castration, an androgen deprivation state may develop in a variety of other ways, including medical castration to inhibit androgen biosynthesis, as well as the use of antiandrogens that block the action of these hormones. This paper aims to review the structure, function, and pathophysiology of the androgen receptor and discuss the role that androgen deprivation therapy plays in the emergence of Castration-Resistant Prostate Cancer (CRPC) neuroendocrine carcinoma and other morphologic variants [3].
The androgens
Androgens include a multitude of hormones synthesized in the testes and adrenal glands. These hormones control and regulate the development and maintenance of male characteristics. Thus, the androgens control and modulate the embryological development of the primary male sex organs and play a crucial role in transforming male secondary sex characteristics during puberty. The principal androgen in males is testosterone, synthesized by the Leydig cells in the testis. The main subsets of adrenal androgens, including androstenedione and Dehydroepiandrosterone (DHEA), are produced in the innermost layer of the adrenal cortex, namely the zona reticularis. Only 1-2% of testosterone in the serum is present in a free state, while the rest is bound to sex hormone-binding globulin. After entering prostate cells, testosterone interacts with 5-alpha-reductase and converts to dihydroxy testosterone, a hormone with a high affinity for binding to AR. Within the prostatic cells, adrenal androgens are also changed to testosterone following interaction with 17-beta-hydroxysteroid dehydrogenase within the prostate cells [4].
Structure and function of androgen receptors
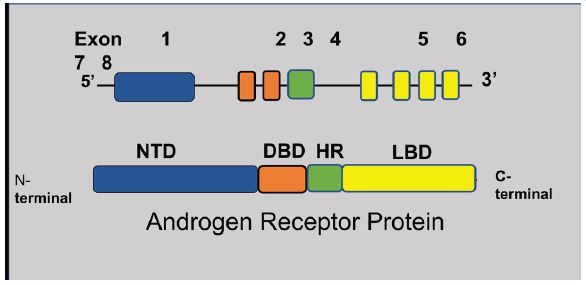
AR has a structure like other hormone receptors for estrogen, progesterone, and glucocorticoids. The gene for AR is located on chromosome X (Xq11-12) and consists of 8 exons that code a protein with approximately 919 amino acids [5,6]. AR includes four regions, namely, an NH2 terminal Transactivation Domain (NTD), a DNA-binding domain (DBD), a hinge, and a Ligand-Binding Domain (LBD) that are encoded by exons 1-6, as depicted in Figure 1.
The NTD contains a polymorphic repeat sequence of cytosine, adenine, and glutamine (CAG repeats). These repeats are of varying length ranging from 8-31, with most men having 19-25 repeats. Shorter repeats are associated with higher transcriptional activity of AR and an increased risk of prostate cancer [6,7]. The availability of androgens and the presence of a functioning AR are crucial for normal male sexual differentiation. In male fetuses a complete loss of a functioning AR is associated with complete androgen insensitivity syndrome [8].
Immunohistochemical analysis of prostatic tissue reveals strong cytoplasmic staining for AR in luminal cells, fibromuscular stromal cells, and endothelial cells while the basal cells are only weakly reactive. The function of androgens is exerted by binding to AR [9]. In normal prostate glandular epithelial cells, AR activation produces secretory proteins, such as prostate-specific antigens (PSAs). AR in inactive state is present in the cytoplasm along with heat shock proteins 70 and 90 that act as chaperone proteins [10]. The androgens, including testosterone and dihydrotestosterone (DHT), exert their function by binding to the ligand-binding domain of AR following which the AR undergoes a conformational change, enters the nucleus, forms a dimer. The AR dimer binds to the androgen-response element of targeted genes resulting in appropriate gene expression (Figure 2). Androgen receptor cofactors consisting of a group of p160 proteins, namely SRC-1, SRC-2, and SRC-3 modulate this action [6,11,12].
Virtually total dependence of prostate cancer on AR
Many cancers acquire key mutations crucial in maintaining proliferative activity in the tumor. Prostate cancers, on the other hand, have a low mutational burden compared to other cancer types. Several sequencing studies on early localized prostate cancers and advanced late-stage disease have demonstrated very few mutations. Thus, this cancer is unique because it has a relatively low mutational burden and entirely depends on the activation signal from the androgen receptor for proliferation, progression, and self-renewal [13,14].
Surgical and medical castration can eliminate or significantly reduce the amount of androgen produced by the testes. However, small amounts of androgens produced by the adrenal may sustain tumor growth and progression. In recent years, several potent non-steroidal antiandrogens such as Flutamide (Eulebicalutamide (Casodex) and nilutamide (Nilandron) have been available. These agents neutralize residual androgen activity as they bind to the AR but do not activate its transcriptional activity. Instead, they competitively block testosterone from binding to the AR [15,16].
Survival of prostate cancer patients has improved with the use of antiandrogens; however, resistance to androgen deprivation therapy develops in most prostate cancers as they become androgen-independent or Castration-Resistant Cancers (CRPC) [17].
Survival strategies of CRPC
Transcriptionally active ARs still drive most CRPCs which can grow under low androgen levels. Several mechanisms may be involved in the development and continued progression of CRPC under these conditions [5,18].
AR gene point mutations
Around 15-30 percent of CPRCs have point mutations in the LBD or NTD region of the AR gene. These mutations cause AR to lose its specificity for testosterone, so other hormones, such as progesterone and estrogen, can also activate the receptor. Even drugs such as flutamide, bicalutamide, and enzalutamide can interact similarly with AR.
Androgen receptor gene amplification
Approximately 30% to 50% of CRPCs may develop amplification of the AR gene. Marked overexpression of AR in these tumors may result in progression even with deficient levels of androgen under androgen deprivation therapy.
Changes in the production of androgen
Androgen deprivation therapy results in a lack of testosterone produced by the testes, although adrenal glands continue to produce small amounts of androgens. Normal prostatic cells and prostatic carcinoma cells may convert these into testosterone, a process that may be markedly enhanced in CRPC.
Altered activity of AR cofactors
Studies on patients with prostate cancer have revealed a significant role of several AR Cofactors in the development and progression of these cancers. Overexpression of AR cofactors may increase transcriptional activity and enhance cellular proliferation signal.
Androgen receptor variants
Many of the over 20 splicing variants of AR may be functionally active even without androgens because they lack the C-terminal domain.
Phenotypic plasticity and development of additional subtypes of prostatic cancer
Approximately a quarter of prostate cancers may become resistant to androgen deprivation therapy without any alterations in AR. The presumed mechanism underlying this change is phenotypic plasticity, which enables the cancer cells to undergo many non-genetic phenotypic cell state changes. These changes amplify the heterogeneity of cancer cells and allow them to acquire migratory, invasive, and chemo-resistant properties. Prostate cancer cells generally have epithelial phenotype and depend on androgen receptor signaling for growth and proliferation. Some tumors may progress to mesenchymal or neuroendocrine phenotypes that no longer require androgen receptor activation for proliferation and growth. Many transcription factors and signaling molecules the tumor cells produce are responsible for these phenotypic changes. These factors are typically involved in lineage programming during embryologic development. However, when expressed in prostate cancer, they induce lineage plasticity and reprogramming that promote resistance to hormonal therapy and increase the tendency for metastasis. No effective treatments are currently available to inhibit or reverse the tumor progression of prostate cancers that use mechanisms of linear plasticity for growth and progression [19-21].
Neuroendocrine carcinomas of the prostate
Normal prostate and prostatic adenocarcinomas contain small neuroendocrine cells randomly distributed among luminal and basal cells. These cells secret various peptide hormones that affect adjacent epithelial cells by paracrine activity. These cells, however, are not the source of neuroendocrine carcinomas of the prostate. With the use of androgen deprivation therapy, new tumor types have emerged that employ multiple AR-independent mechanisms for survival. A subset of CRPC tumors partially or entirely lose androgen receptor activity and PSA expression. These tumors employ tumor plasticity and acquire neuroendocrine phenotype for further progression and dissemination. Thus, these tumors are nonreactive or only focally positive for AR and PSA but manifest positive staining for neuroendocrine markers, including chromogranin, CD-56, synaptophysin, and neuron-specific enolase. Post-therapy neuroendocrine carcinomas may develop in 10-17% of CRPC. These carcinomas have low or absent androgen receptors and are not responsive to therapies that target AR signaling [22].
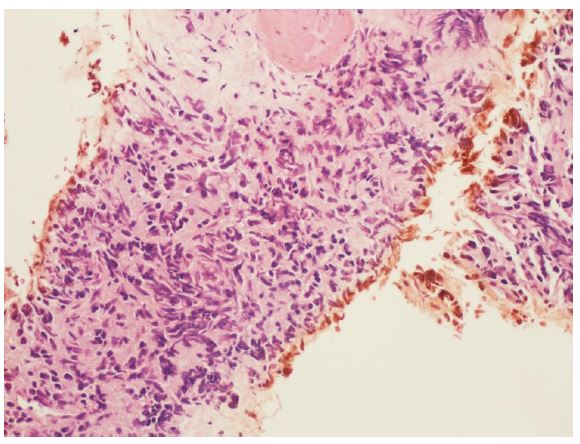
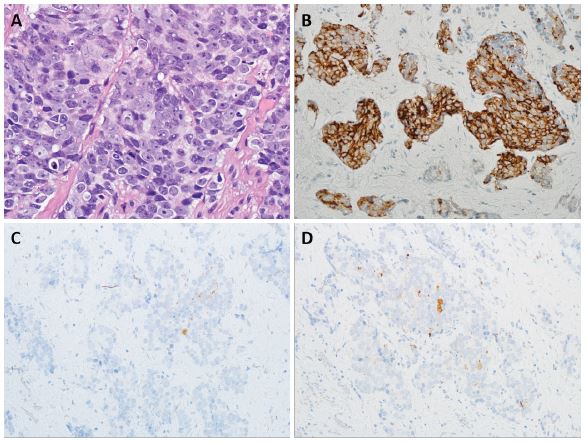
Small cell carcinoma is the most common neuroendocrine carcinoma of the prostate [23] (Figure 3). The morphology of this tumor is identical to small cell carcinoma of the lung. The tumor cells have scanty cytoplasm, slightly elongated hyperchromatic, molded nuclei with inconspicuous nucleoli, and a high proliferation rate. These tumors frequently show necrosis and crushing artifacts. In rare cases, the neoplasm may resemble a carcinoid tumor. Another uncommon variant is large-cell neuroendocrine carcinoma, with cells containing abundant cytoplasm and large pleomorphic nuclei frequently containing prominent nucleoli [24] (Figure 4). Rarely, the carcinoma cells depict large cell morphology but reveal a dual pattern of immunohistochemical staining where the tumor cells express neuroendocrine markers but also stain for AR and PSA [25]. These tumors may represent an intermediate stage of transdifferentiation from prostatic adenocarcinoma to neuroendocrine carcinoma and have been variably described as amphicrine carcinoma and large cell carcinoma with diffuse neuroendocrine differentiation [24,25]. This entity, however, still needs to be defined. Tumors with Paneth cell-like cytoplasmic granules are another uncommon variant of neuroendocrine carcinoma of the prostate. The prognosis of these tumors is determined by accompanying conventional adenocarcinoma component [24,26-28]. Rare cases of prostate cancer lacking both AR and neuroendocrine expression may be seen. These tumors may have variable morphologic features, including squamous, spindle cell, and small cell components [29,30].
De novo neuroendocrine carcinomas of the prostate are much less common, representing less than 2% of the prostatic carcinomas. These tumors occur in patients lacking any history of cancer therapy and have similar morphology to that of post-therapy neuroendocrine carcinoma, including small-cell, large-cell, and carcinoid-like patterns. The patients with these tumors usually present with an advanced disease and have a higher mortality rate [31,32]. The neuroendocrine features may be present in the primary location, metastatic site, or both [33].
Therapies targeting androgen receptor in prostate cancer: Current status and future directions
Following androgen deprivation therapy, prostate cancers usually relapse as they evolve into CRPCs with a strong tendency to develop metastatic disease. In the last decade, crucial advances in the treatment of prostate cancer have been made, and novel treatment strategies have been developed. The androgen receptor has emerged as the most crucial factor regulating the activity and spread of cancer cells. Consequently, most current treatment regimens are designed to block or modify the AR pathway. These therapies help to reduce androgen production and suppress AR activity. Unfortunately, these treatment modalities provide only temporary tumor suppression in CRPC patients and fail to produce a complete cure. This lack of curative relief is due to various factors, including AR gene mutations or splicing variations, which result in AR reactivation. Complete elimination of the AR protein in prostate cancer cells is a promising solution that can provide curative relief. Multiple strategies have emerged, and several potent agents that reduce AR protein levels were reported. The potential mechanisms involved include inhibition of heat-shock proteins, suppression of AR splicing, prevention of nuclear localization of AR, and suppression of AR N-terminal, among others [34-40]. Using small molecules to block AR dimerization and thus prevent its activation is another promising strategy against prostate cancer [41]. Thus, the elimination of AR protein or its function appears to be a realistic solution for avoiding AR reactivation during androgen deprivation therapy in prostate cancers.
Conclusion
In conclusion, this paper briefly updates the structure and function of the androgen receptor and reviews its role in the development and progression of prostate cancer. The androgen receptor is currently an essential target for prostate cancer therapy.
References
- Feng Q, He B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Frontiers in Oncology. 2019; 9. https://doi.org/10.3389/fonc.2019.00858
- Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941; 1: 293-297
- ES, Cil E, Brooke GN. Use of Antiandrogens as Therapeutic Agents in COVID-19 Patients. Viruses. 2022; 14(12): 2728. doi: 10.3390/v14122728.
- Patrão MT, Silva EJ, Avellar MC. Androgens and the male reproductive tract: An overview of classical roles and current perspectives. Arq Bras Endocrinol Metabol. 2009; 53(8): 934-45. doi: 10.1590/s0004-27302009000800006.
- Tan M, Li J, Xu H, et al. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015; 36: 3-23. https://doi.org/10.1038/aps.2014.18
- Fujita K, Nonomura N. Role of Androgen Receptor in Prostate Cancer: A Review. World J Mens Health. 2019; 37(3): 288-295. doi: 10.5534/wjmh.180040.
- Qin Z, Li X, Han P, Zheng Y, Liu H, et al. Association between polymorphic CAG repeat lengths in the androgen receptor gene and susceptibility to prostate cancer: A systematic review and meta-analysis. Medicine (Baltimore). 2017; 96(25): e7258. doi: 10.1097/MD.0000000000007258.
- Ieuan A Hughes, John D Davies, Trevor I Bunch, Vickie Pasterski, Kiki Mastroyannopoulou. Jane MacDougall Androgen insensitivity syndrome. Lancet. 2012; 380: 1419-28.
- Chodak GW, Kranc DM, Puy LA, Takeda H, Johnson K, Chang C. Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate. J Urol. 1992; 147(3 Pt 2): 798-803. doi: 10.1016/s0022-5347(17)37389-5.
- Albany C, Hahn NM. Heat shock and other apoptosis-related proteins as therapeutic targets in prostate cancer. Asian Journal of Andrology. 2014; 16(3): 359-363. https://doi.org/10.4103/1008-682X.126400
- Xu J, Wu C. Normal and Cancer-Related Functions of the p160 Steroid Receptor Coactivator (SRC) Family. Nature reviews. Cancer. 2009; 9: 615. https://doi.org/10.1038/nrc2695
- Zamagni A, Cortesi M, Zanoni M, Tesei A. Non-nuclear AR Signaling in Prostate Cancer. Frontiers in Chemistry. 2019; 7. https://doi.org/10.3389/fchem.2019.00651
- Chida K, Kawazoe A, Kawazu M, Suzuki T, Nakamura Y, et al. A Low Tumor Mutational Burden and PTEN Mutations Are Predictors of a Negative Response to PD-1 Blockade in MSI-H/dMMR Gastrointestinal Tumors. Clin Cancer Res. 2021 Jul 1;27(13):3714-3724. doi: 10.1158/1078-0432.CCR-21-0401. Epub 2021 Apr 29. PMID: 33926917.
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015 Nov 5;163(4):1011-25. doi: 10.1016/j.cell.2015.10.025. PMID: 26544944; PMCID: PMC4695400.
- Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013 Nov;154(11):4010-7. doi: 10.1210/en.2013-1466. Epub 2013 Sep 3. PMID: 24002034; PMCID: PMC3948917.
- Eva Estébanez-Perpiñá 1 , Charlotte L. Bevan 2 and Iain J. McEwan 3,*Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes on Cancers 2021, 13, 509. https://doi.org/10.3390/cancers13030509.
- Morote J, Aguilar A, Planas J, Trilla E. Definition of Castrate Resistant Prostate Cancer: New Insights. Biomedicines. 2022 Mar 17;10(3):689. doi: 10.3390/biomedicines10030689. PMID: 35327491; PMCID: PMC8945091.
- Sharifi, N. (2013). Mechanisms of Androgen Receptor Activation in Castration-Resistant Prostate Cancer. Endocrinology, 154(11), 4010-4017. https://doi.org/10.1210/en.2013-1466
- Nanda JS, Koganti P, Perri G, Ellis L. Phenotypic Plasticity - Alternate Transcriptional Programs Driving Treatment Resistant Prostate Cancer. Crit Rev Oncog. 2022;27(1):45-60. doi: 10.1615/CritRevOncog.2022043096. PMID: 35993978.
- Jolly, M. K., Kulkarni, P., Weninger, K., Orban, J., & Levine, H. (2018). Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Frontiers in Oncology, 8. https://doi.org/10.3389/fonc.2018.00050
- Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol. 2018 May;15(5):271-286. doi: 10.1038/nrurol.2018.22. Epub 2018 Feb 20. PMID: 29460922.
- Patel GK, Chugh N, Tripathi M. Neuroendocrine Differentiation of Prostate Cancer-An Intriguing Example of Tumor Evolution at Play. Cancers (Basel). 2019 Sep 20;11(10):1405. doi: 10.3390/cancers11101405. PMID: 31547070; PMCID: PMC6826557.
- Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, Robinson BD, Troncoso P, Rubin MA. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol. 2014 Jun;38(6):756-67. doi: 10.1097/PAS.0000000000000208. PMID: 24705311; PMCID: PMC4112087
- Fine, S. Neuroendocrine tumors of the prostate. Mod Pathol 31 (Suppl 1), 122–132 (2018). https://doi.org/10.1038/modpathol.2017.164
- Prendeville S, Al-Bozom I, Compérat E, Sweet J, Evans AJ, Ben-Gashir M, Mete O, van der Kwast TH, Downes MR. Prostate carcinoma with amphicrine features: further refining the spectrum of neuroendocrine differentiation in tumours of primary prostatic origin? Histopathology. 2017 Dec;71(6):926-933. doi: 10.1111/his.13330. Epub 2017 Oct 6. PMID: 28756619
- Weaver MG, Abdul-Karim FW, Srigley J, Bostwick DG, Ro JY, Ayala AG. Paneth cell–like change of the prostate gland. A histological, immunohistochemical, and electron microscopic study. Am J Surg Pathol. 1992; 16:62–8.
- Park K, Chen Z, MacDonald TY, Siddiqui J, Ye H, Erbersdobler A, Shevchuk MM, Robinson BD, Sanda MG, Chinnaiyan AM, Beltran H, Rubin MA, Mosquera JM. Prostate cancer with Paneth cell-like neuroendocrine differentiation has recognizable histomorphology and harbors AURKA gene amplification. Hum Pathol. 2014 Oct;45(10):2136-43. doi: 10.1016/j.humpath.2014.06.008. Epub 2014 Jun 26. PMID: 25128228; PMCID: PMC4414025.
- So JS, Gordetsky J, Epstein JI. Variant of prostatic adenocarcinoma with Paneth cell-like neuroendocrine differentiation readily misdiagnosed as Gleason pattern 5. Hum Pathol. 2014 Dec;45(12):2388-93. doi: 10.1016/j.humpath.2014.08.004. Epub 2014 Aug 23. PMID: 25277321.
- Acosta-Gonzalez G, Qin J, Wieczorek R, Melamed J, Deng FM, Zhou M, Makarov D, Ye F, Pei Z, Pincus MR, Lee P. De novo large cell neuroendocrine carcinoma of the prostate, case report and literature review. Am J Clin Exp Urol. 2014 Dec 25;2(4):337-42. PMID: 25606580; PMCID: PMC4297330.
- Cackowski FC, Kumar-Sinha C, Mehra R, Wu YM, Robinson DR, Alumkal JJ, Chinnaiyan AM. Double-Negative Prostate Cancer Masquerading as a Squamous Cancer of Unknown Primary: A Clinicopathologic and Genomic Sequencing-Based Case Study. JCO Precis Oncol. 2020 Nov 16;4:PO.20.00309. doi: 10.1200/PO.20.00309. PMID: 33283137; PMCID: PMC7713564.
- Sawazaki H, Asano A, Kitamura Y, Katsuta J, Ito Y. Androgen receptor-neuroendocrine double-negative tumor with squamous differentiation arising from treatment-refractory metastatic castration-resistant prostate cancer. IJU Case Rep. 2021 Aug 22;4(6):417-420. doi: 10.1002/iju5.12363. PMID: 34755072; PMCID: PMC8560456.
- Abdulfatah, E., Reichert, Z.R., Davenport, M.S. et al. De novo neuroendocrine transdifferentiation in primary prostate cancer–a phenotype associated with advanced clinico-pathologic features and aggressive outcome. Med Oncol 38, 26 (2021). https://doi.org/10.1007/s12032-021-01473-2
- Gopalan A, Al-Ahmadie H, Chen YB, Sarungbam J, Sirintrapun SJ, Tickoo SK, Reuter VE, Fine SW. Neuroendocrine differentiation in the setting of prostatic carcinoma: contemporary assessment of a consecutive series. Histopathology. 2022 Aug;81(2):246-254. doi: 10.1111/his.14707. Epub 2022 Jul 11. PMID: 35758203; PMCID: PMC9327588.
- Ehsani M, David FO, Baniahmad A. Androgen Receptor-Dependent Mechanisms Mediating Drug Resistance in Prostate Cancer. Cancers. 2021; 13(7):1534. https://doi.org/10.3390/cancers13071534
- Le TK, Duong QH, Baylot V, Fargette C, Baboudjian M, Colleaux L, Taïeb D, Rocchi P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers (Basel). 2023 Oct 19;15(20):5047. doi: 10.3390/cancers15205047. PMID: 37894414; PMCID: PMC10605314.
- Le TK, Duong QH, Baylot V, Fargette C, Baboudjian M, Colleaux L, Taïeb D, Rocchi P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers (Basel). 2023 Oct 19;15(20):5047. doi: 10.3390/cancers15205047. PMID: 37894414; PMCID: PMC10605314.
- Hwang, M. W., & Aragon-Ching, J. B. (2023). Mechanistic Insights on Localized to Metastatic Prostate Cancer Transition and Therapeutic Opportunities. Research and Reports in Urology, 15, 519-529. https://doi.org/10.2147/RRU.S386517
- Mehralivand S, Thomas C, Puhr M, Claessens F, van de Merbel AF, Dubrovska A, Jenster G, Bernemann C, Sommer U, Erb HHH. New advances of the androgen receptor in prostate cancer: report from the 1st International Androgen Receptor Symposium. J Transl Med. 2024 Jan 18;22(1):71. doi: 10.1186/s12967-024-04878-5. PMID: 38238739; PMCID: PMC10795409.
- Hahn AW, Siddiqui BA, Leo J, Dondossola E, Basham KJ, Miranti CK, Frigo DE. Cancer Cell-Extrinsic Roles for the Androgen Receptor in Prostate Cancer. Endocrinology. 2023 Apr 17;164(6):bqad078. doi: 10.1210/endocr/bqad078. PMID: 37192413; PMCID: PMC10413433.
- S. Thomas, C., Puhr, M. et al. New advances of the androgen receptor in prostate cancer: report from the 1st International Androgen Receptor Symposium. J Transl Med 22, 71 (2024). https://doi.org/10.1186/s12967-024-04878-5
- Gao H, Zhang JY, Zhao LJ, Yuan-Yuan Guo YY Synthesis and application of clinically approved small-molecule drugs targeting androgen receptor. Bioorganic Chemistry Volume 143, February 2024, 106998.